

Early mitochondrial dysfunction in long-lived Mclk1+/- mice
- PMID: 18635541
- PMCID: PMC3258865
- DOI: 10.1074/jbc.M803287200
Early mitochondrial dysfunction in long-lived Mclk1+/- mice
Abstract
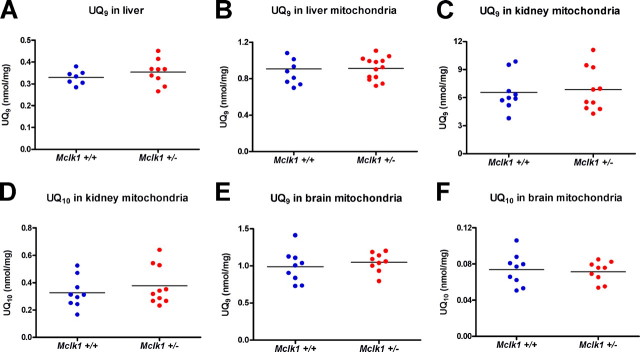
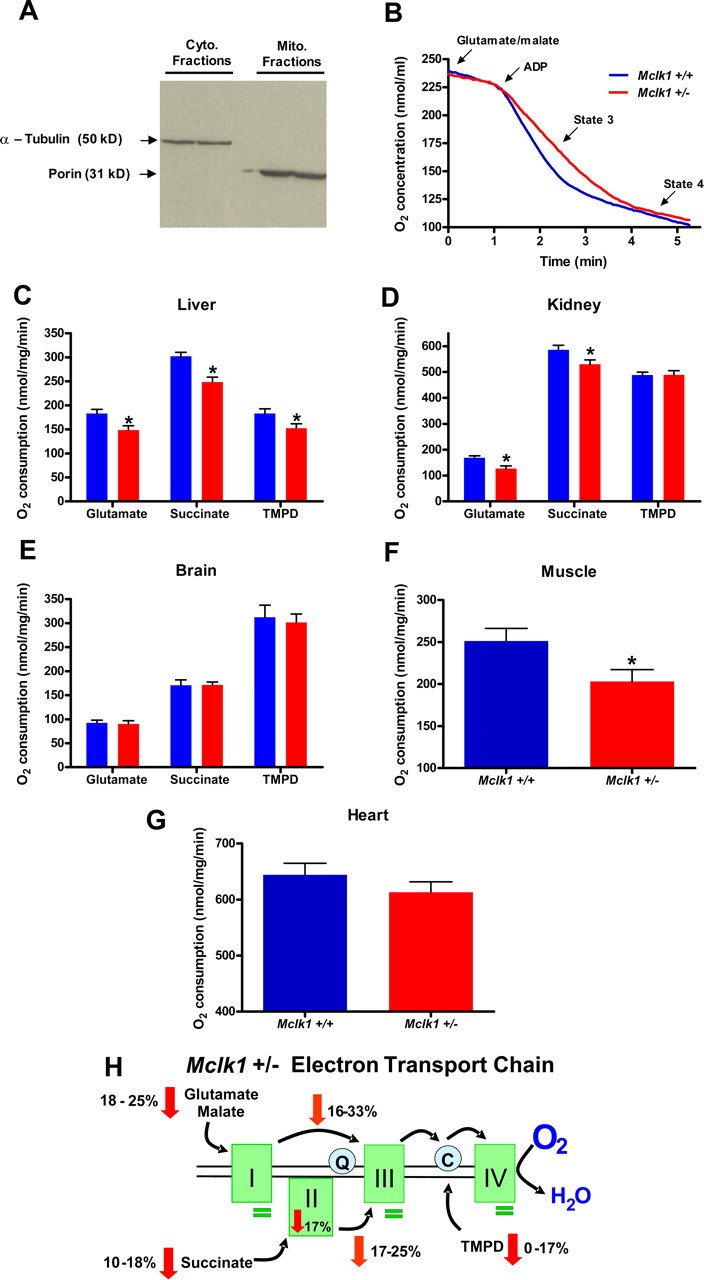
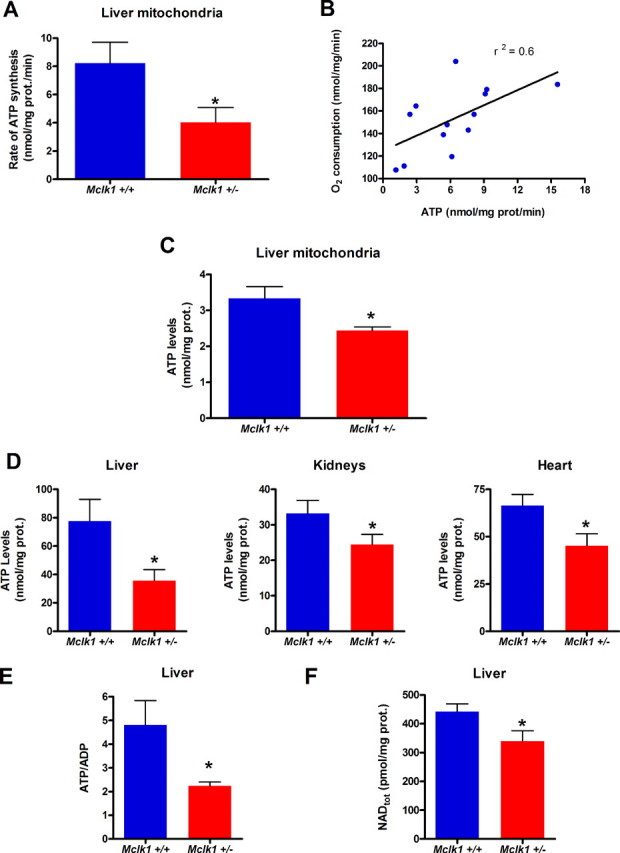
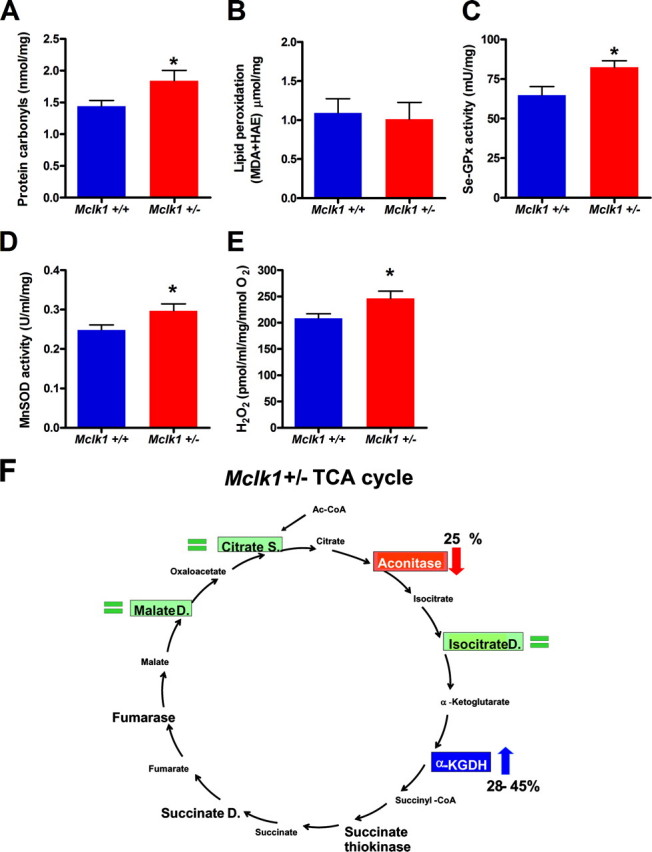
Reduced activity of CLK-1/MCLK1 (also known as COQ7), a mitochondrial enzyme that is necessary for ubiquinone biosynthesis, prolongs the lifespan of nematodes and mice by a mechanism that is distinct from that of the insulin signaling pathway. Here we show that 2-fold reduction of MCLK1 expression in mice reveals an additional function for the protein, as this level of reduction does not affect ubiquinone levels yet affects mitochondrial function substantially. Indeed, we observe that the phenotype of young Mclk1(+/-) mutants includes a severe reduction of mitochondrial electron transport, ATP synthesis, and total nicotinamide adenine dinucleotide (NAD(tot)) pool size as well as an alteration in the activity of key enzymes of the tricarboxylic acid cycle. Surprisingly, we also find that Mclk1 heterozygosity leads to a dramatic increase in mitochondrial oxidative stress by a variety of measures. Furthermore, we find that the mitochondrial dysfunction is accompanied by a decrease in oxidative damage to cytosolic proteins as well as by a decrease in plasma isoprostanes, a systemic biomarker of oxidative stress and aging. We propose a mechanism for the conjunction of low ATP levels, high mitochondrial oxidative stress, and low non-mitochondrial oxidative damage in a long-lived mutant. Our model helps to clarify the relationship between energy metabolism and the aging process and suggests the need for a reformulation of the mitochondrial oxidative stress theory of aging.
Figures
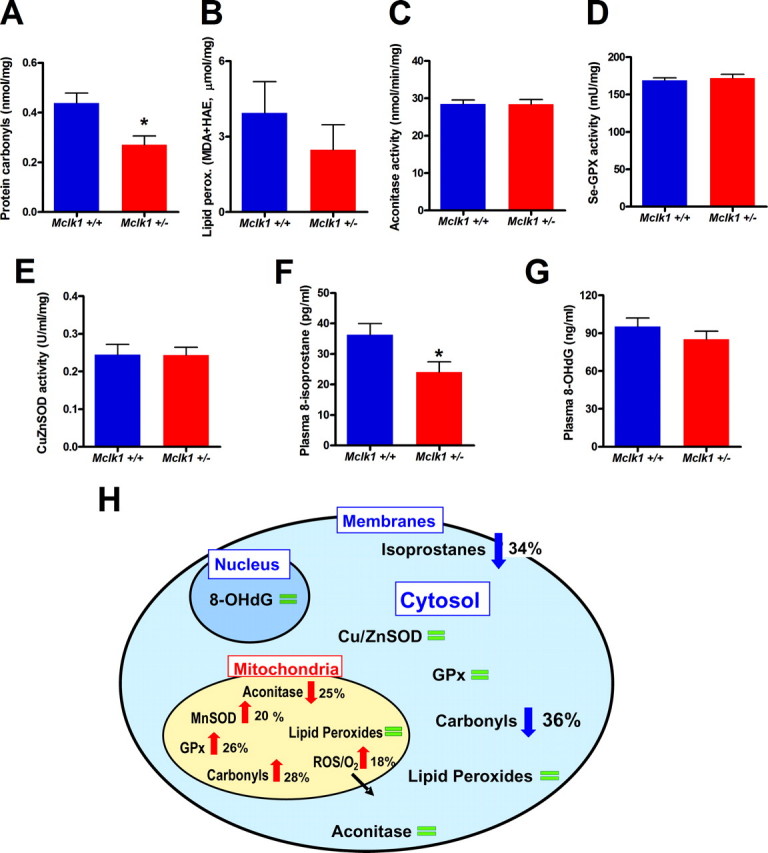
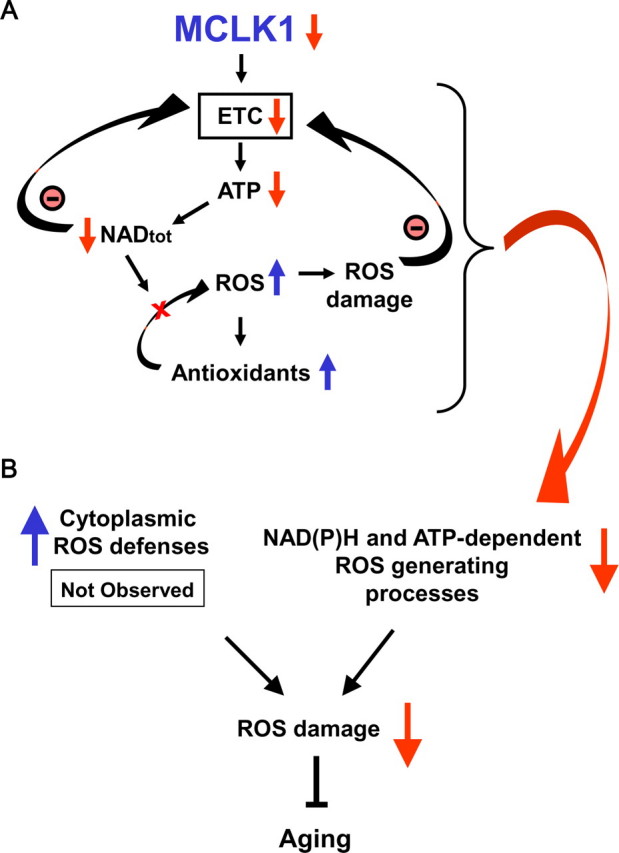
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
