

doi: 10.1016/j.jprot.2008.07.003.
Epub 2008 Jul 12.
Protein identification pipeline for the homology-driven proteomics
Affiliations
- PMID: 18639657
- PMCID: PMC2644553
- DOI: 10.1016/j.jprot.2008.07.003
Item in Clipboard
Protein identification pipeline for the homology-driven proteomics
J Proteomics.
.
Abstract
Homology-driven proteomics is a major tool to characterize proteomes of organisms with unsequenced genomes. This paper addresses practical aspects of automated homology-driven protein identifications by LC-MS/MS on a hybrid LTQ Orbitrap mass spectrometer. All essential software elements supporting the presented pipeline are either hosted at the publicly accessible web server, or are available for free download.
Figures

The output of PepNovo batch mode interpretation of MS/MS spectra from LC-MS/MS run. For each interpreted spectrum PepNovo provides: neutral mass of the peptide precursor (a); spectrum name, including the precursor charge state (b); total ion current (TIC) in MS/MS spectrum (c); TIC fraction covered by expected fragments of the top candidate sequence (d); sequence quality score representing the expected number of correct amino acids in the top candidate sequence (e); candidate sequences (f), formatted according to MS BLAST conventions: B = R or K (generic trypsin cleavage site); Z = Q or K (if indistinguishable in low resolution MS/MS spectra); L = L or I; M+15.99 = methionine sulfoxide residue; X = undetermined amino acid residue.

Web interface of the MS BLAST server. LC-MSMS Presets check box activates the settings that make possible MS BLAST searches with large peptide sequence queries produced by automated interpretation of MS/MS spectra acquired by data-dependent LC-MS/MS. A part of the search string is shown in the submission window. MS BLAST utilizes degenerate, redundant and partially accurate sequence queries. Usually, up to 7 peptide sequence candidates per each interpreted MS/MS spectrum are included into the search query. Precursor masses, scan numbers, sequence quality scores and other parameters simplifying handling of de novo output, are ignored by MS BLAST server. MS BLAST server can process a query comprising up to 150,000 amino acid residues, which is formally equivalent to BLAST search with the sequence of ca. 16.5 megadalton protein chimera.
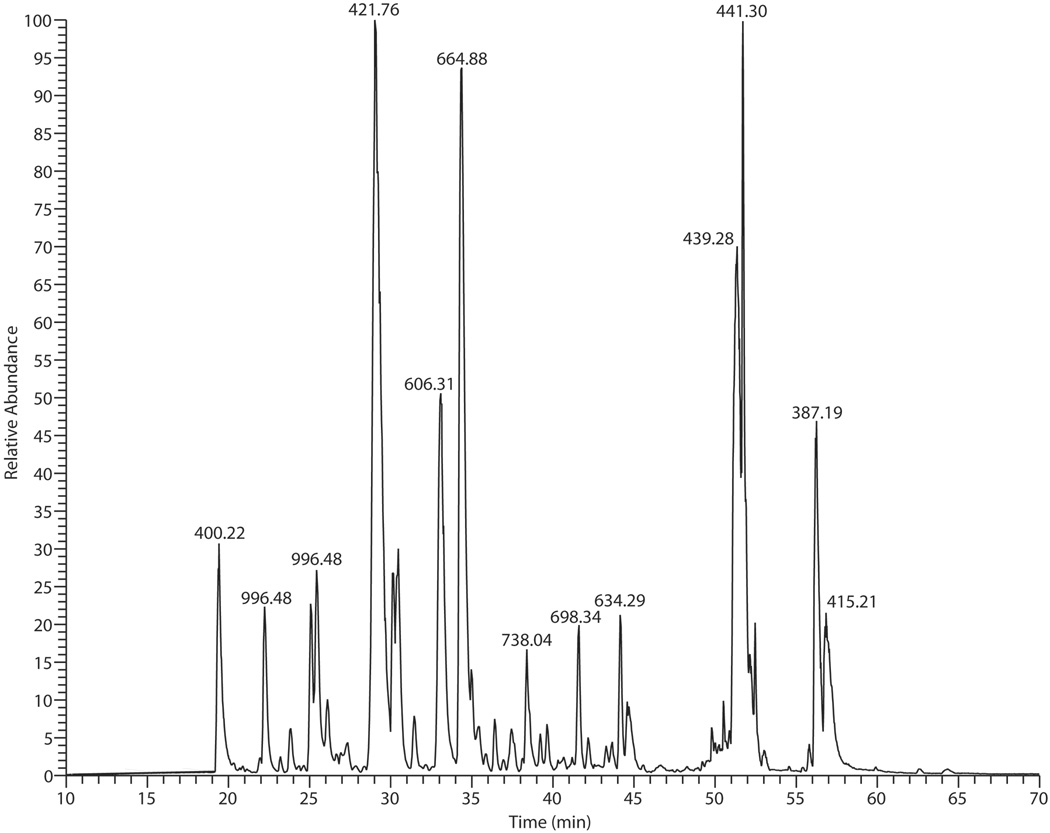
Base peak LC-MS/MS chromatogram of in-gel tryptic digest of a silver stained spot with apparent MW of 19 kDa and pI of 8.0 excised from a 2D gel of Triatoma infestans saliva. The analysis produced, in total, 2210 MS/MS spectra acquired from doubly- and triply- charged precursor ions. Peaks at the chromatogram are designated with base peak m/z.

De novo interpretation of MS/MS spectrum of the precursor ion with m/z 922.933 by PepNovo software. The interpretation of the spectrum (panel A) acquired on a linear ion trap analyzer produced several candidate sequences (inset B) with the sequence quality score of the top candidate of 13.6. Along with candidate sequences from other fragmented precursors, they were submitted to MS BLAST search that produced the sequence alignment presented in Table 1. Peaks in the spectrum (panel A) are designated according to the fragment type and m/z, computed from the aligned peptide sequence.
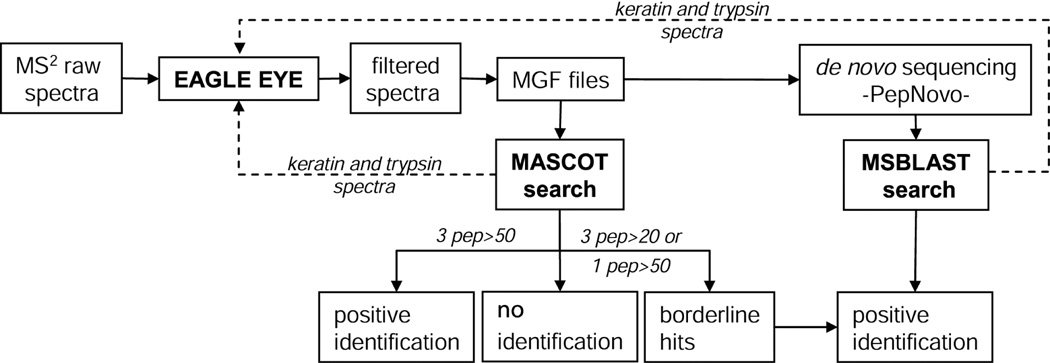
Protein identification workflow that uses a combination of MASCOT searches and de novo sequencing followed by MS BLAST searches. First, all spectra were filtered against a background spectra library by EagleEye software, which removed a large number of background MS/MS spectra irrespective of their quality, annotation and origin. Filtered data file in .mgf format was submitted to MASCOT searches against a comprehensive MSDB database. If 3 or more unique peptides were matched by MS/MS spectra with ions score exceeding 50, these identification were considered positive. If 3 peptides were matched with ions scores above 20 but below 50, or only one peptide was matched with the score above 50, hits were considered borderline and subjected to further validation by de novo sequencing. In parallel, the same .mgf file with filtered spectra was subjected to batch de novo sequencing followed by MS BLAST search with obtained candidate sequences. For identifications solely based on sequence similarity and for independent validation of MASCOT borderline hits, MS BLAST scoring scheme was applied.
References
-
- Forner F, Foster LJ, Toppo S. Mass spectrometry data analysis in the proteomics era. Current Bioinformatics. 2007;2:63–93.
-
- Sadygov RG, Liu H, Yates JR. Statistical models for protein validation using tandem mass spectral data and protein amino acid sequence databases. Anal Chem. 2004;76:1664–1671. - PubMed
-
- Nesvizhskii AI, Aebersold R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Discov Today. 2004;9:173–181. - PubMed
-
- Liska AJ, Shevchenko A. Expanding organismal scope of proteomics: cross-species protein identification by mass spectrometry and its implications. Proteomics. 2003;3:19–28. - PubMed
-
- Liska AJ, Shevchenko A. Combining mass spectrometry with database interrogation strategies in proteomics. Trends Anal Chem. 2003;22:291–298.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
