

Backbone relaxation coupled to the ionization of internal groups in proteins: a self-guided Langevin dynamics study
- PMID: 18641078
- PMCID: PMC2567956
- DOI: 10.1529/biophysj.108.130906
Backbone relaxation coupled to the ionization of internal groups in proteins: a self-guided Langevin dynamics study
Abstract
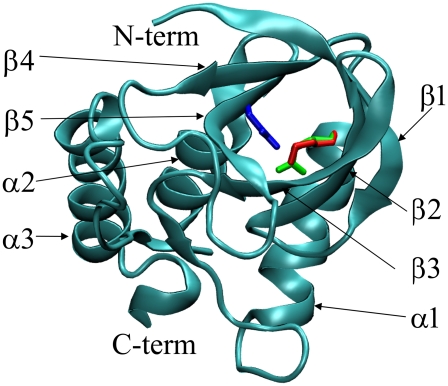
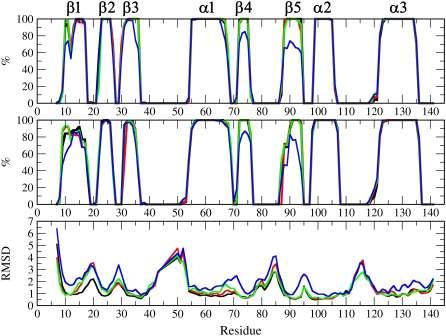
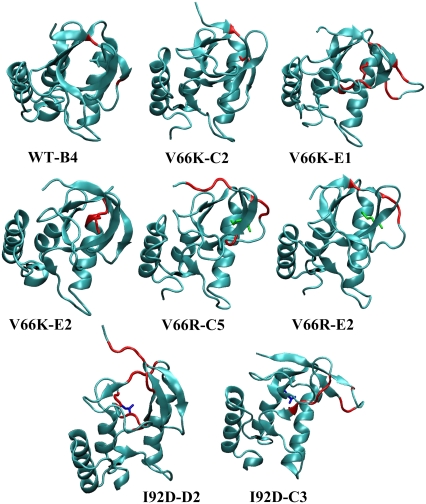
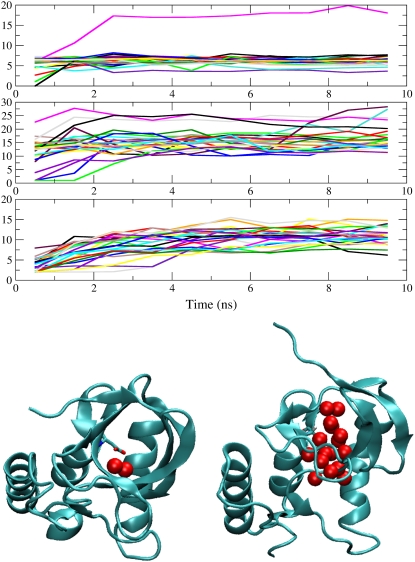
Pathways of structural relaxation triggered by ionization of internal groups in staphylococcal nuclease (SNase) were studied through multiple self-guided Langevin dynamics (SGLD) simulations. Circular dichroism, steady-state Trp fluorescence, and nuclear magnetic resonance spectroscopy have shown previously that variants of SNase with internal Glu, Asp, and Lys at positions 66 or 92, and Arg at position 66, exhibit local reorganization or global unfolding when the internal ionizable group is charged. Except for Arg-66, these internal ionizable groups have unusual pKa values and are neutral at physiological pH. The structural trends observed in the simulations are in general agreement with experimental observations. The I92D variant, which unfolds globally upon ionization of Asp-92, in simulations often exhibits extensive hydration of the protein core, and sometimes also significant perturbations of the beta-barrel. In the crystal structure of the V66R variant, the beta1 strand from the beta-barrel is domain-swapped; in the simulations, the beta1 strand is sometimes partially released. The V66K variant, which in solutions shows reorganization of six residues at the C-terminus of helix alpha1 and perturbations in the beta-barrel structure, exhibits fraying of three residues of helix alpha1 in one simulation, and perturbations and partial unfolding of three beta-strands in a few other simulations. In sharp contrast, very small structural changes were observed in simulations of the wild-type protein. The simulations indicate that charging of internal groups frequently triggers penetration of water into the protein interior. The pKa values of Asp-92 and Arg-66 calculated with continuum methods on SGLD-relaxed structures reached the normal values in most simulations. Detailed analysis of accuracy and performance of SGLD demonstrates that SGLD outperforms LD in sampling of alternative protein conformations without loss of the accuracy and level of detail characteristic of regular LD.
Figures
References
-
- Hoff, W. D., A. Xie, I. H. M. van Stokkum, X.-J. Tang, J. Gural, A. R. Kroon, and K. J. Hellingwerf. 1999. Global conformational changes upon receptor stimulation in photoactive yellow protein. Biochemistry. 38:1009–1017. - PubMed
-
- Lee, B.-C., P. A. Croonquist, T. R. Sosnick, and W. D. Hoff. 2001. PAS domain receptor photoactive yellow protein is converted to a molten globule state upon activation. J. Biol. Chem. 276:20821–20823. - PubMed
-
- Xie, A., L. Keleman, J. Hendriks, B. J. White, K. J. Hellingwerf, and W. D. Hoff. 2001. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 40:1510–1517. - PubMed
-
- Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999. Structure changes in bacteriorhodopsin during ion transport at 2 Å resolution. Science. 286:255–260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
