

PatternLab for proteomics: a tool for differential shotgun proteomics
- PMID: 18644148
- PMCID: PMC2488363
- DOI: 10.1186/1471-2105-9-316
PatternLab for proteomics: a tool for differential shotgun proteomics
Abstract
Background: A goal of proteomics is to distinguish between states of a biological system by identifying protein expression differences. Liu et al. demonstrated a method to perform semi-relative protein quantitation in shotgun proteomics data by correlating the number of tandem mass spectra obtained for each protein, or "spectral count", with its abundance in a mixture; however, two issues have remained open: how to normalize spectral counting data and how to efficiently pinpoint differences between profiles. Moreover, Chen et al. recently showed how to increase the number of identified proteins in shotgun proteomics by analyzing samples with different MS-compatible detergents while performing proteolytic digestion. The latter introduced new challenges as seen from the data analysis perspective, since replicate readings are not acquired.
Results: To address the open issues above, we present a program termed PatternLab for proteomics. This program implements existing strategies and adds two new methods to pinpoint differences in protein profiles. The first method, ACFold, addresses experiments with less than three replicates from each state or having assays acquired by different protocols as described by Chen et al. ACFold uses a combined criterion based on expression fold changes, the AC test, and the false-discovery rate, and can supply a "bird's-eye view" of differentially expressed proteins. The other method addresses experimental designs having multiple readings from each state and is referred to as nSVM (natural support vector machine) because of its roots in evolutionary computing and in statistical learning theory. Our observations suggest that nSVM's niche comprises projects that select a minimum set of proteins for classification purposes; for example, the development of an early detection kit for a given pathology. We demonstrate the effectiveness of each method on experimental data and confront them with existing strategies.
Conclusion: PatternLab offers an easy and unified access to a variety of feature selection and normalization strategies, each having its own niche. Additionally, graphing tools are available to aid in the analysis of high throughput experimental data. PatternLab is available at http://pcarvalho.com/patternlab.
Figures
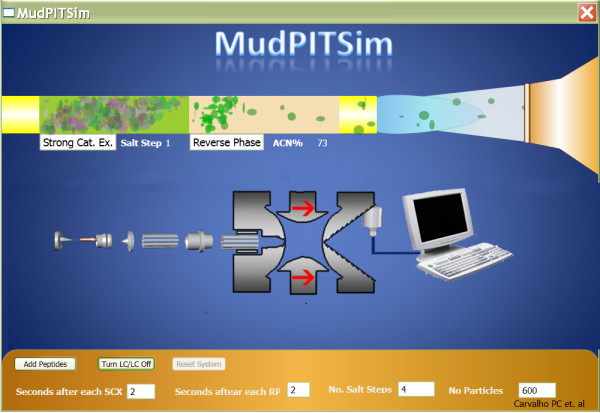
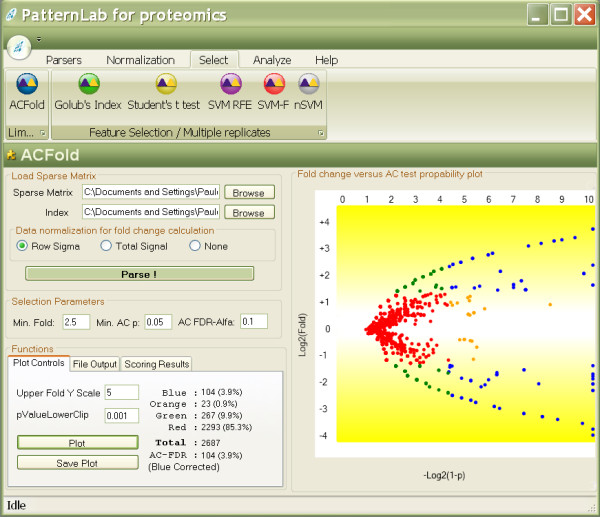
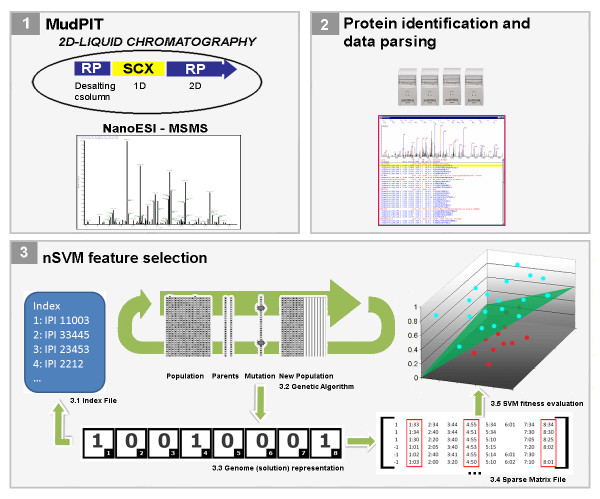
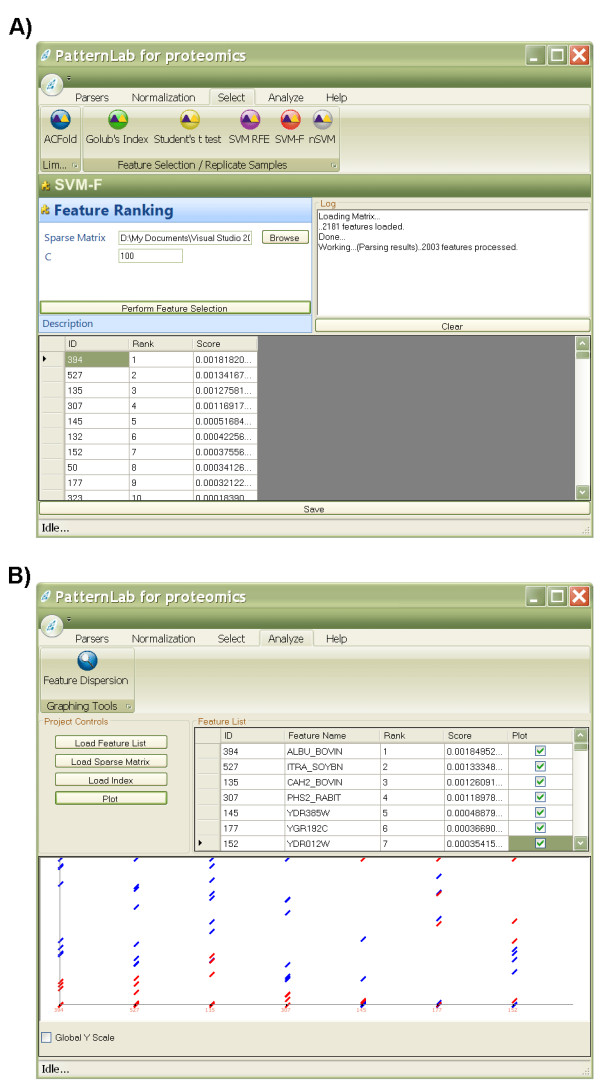
References
-
- Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolster D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–526. doi: 10.1038/nature01107. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
