

Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs
- PMID: 18648507
- PMCID: PMC2447871
- DOI: 10.1371/journal.pone.0002718
Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs
Abstract
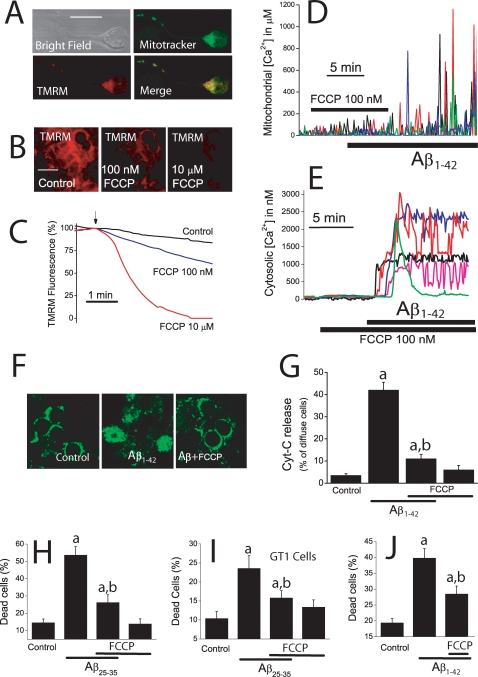
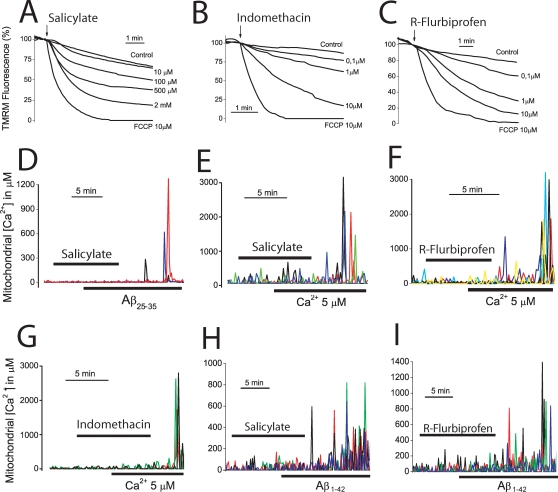
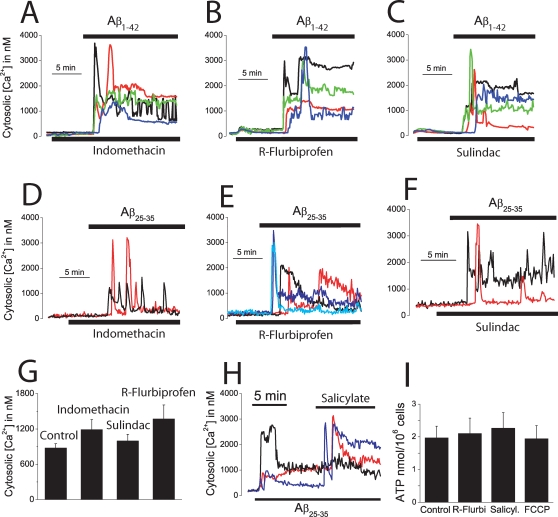
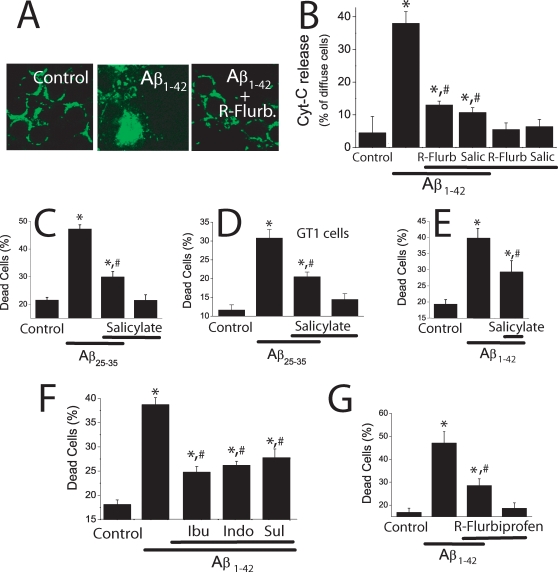
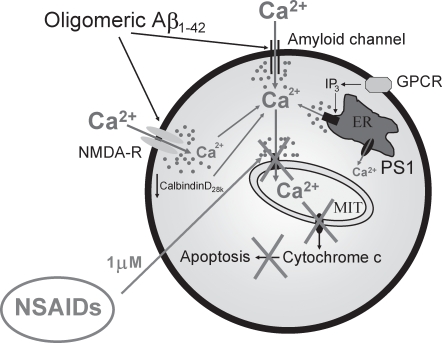
Dysregulation of intracellular Ca(2+) homeostasis may underlie amyloid beta peptide (Abeta) toxicity in Alzheimer's Disease (AD) but the mechanism is unknown. In search for this mechanism we found that Abeta(1-42) oligomers, the assembly state correlating best with cognitive decline in AD, but not Abeta fibrils, induce a massive entry of Ca(2+) in neurons and promote mitochondrial Ca(2+) overload as shown by bioluminescence imaging of targeted aequorin in individual neurons. Abeta oligomers induce also mitochondrial permeability transition, cytochrome c release, apoptosis and cell death. Mitochondrial depolarization prevents mitochondrial Ca(2+) overload, cytochrome c release and cell death. In addition, we found that a series of non-steroidal anti-inflammatory drugs (NSAIDs) including salicylate, sulindac sulfide, indomethacin, ibuprofen and R-flurbiprofen depolarize mitochondria and inhibit mitochondrial Ca(2+) overload, cytochrome c release and cell death induced by Abeta oligomers. Our results indicate that i) mitochondrial Ca(2+) overload underlies the neurotoxicity induced by Abeta oligomers and ii) inhibition of mitochondrial Ca(2+) overload provides a novel mechanism of neuroprotection by NSAIDs against Abeta oligomers and AD.
Conflict of interest statement
Figures
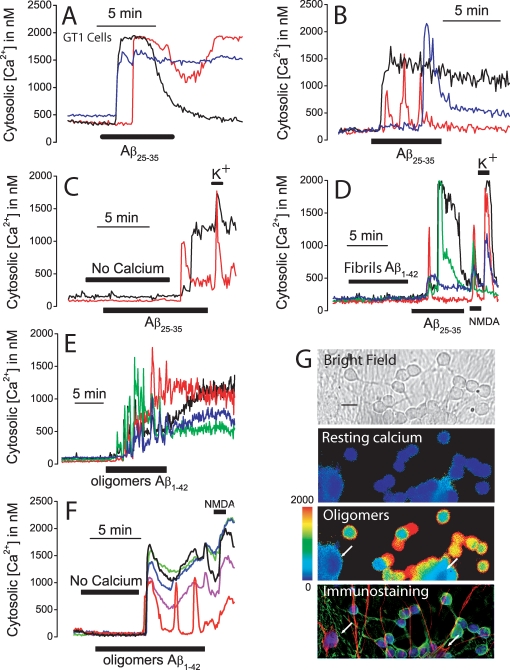
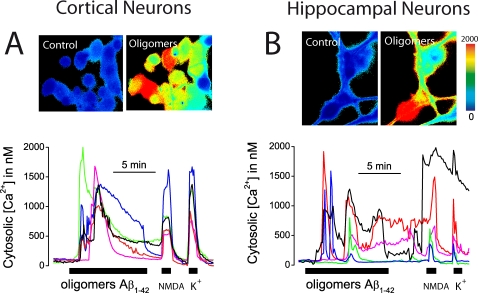
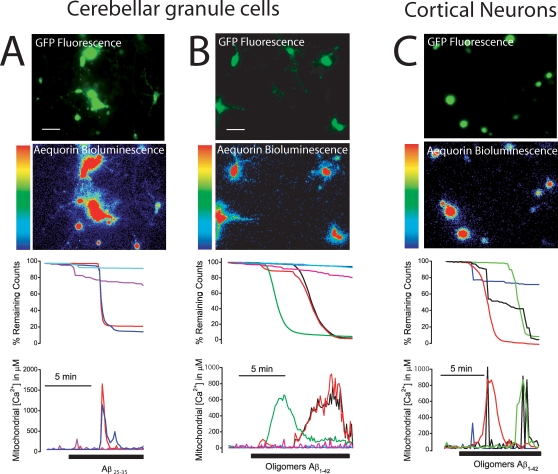
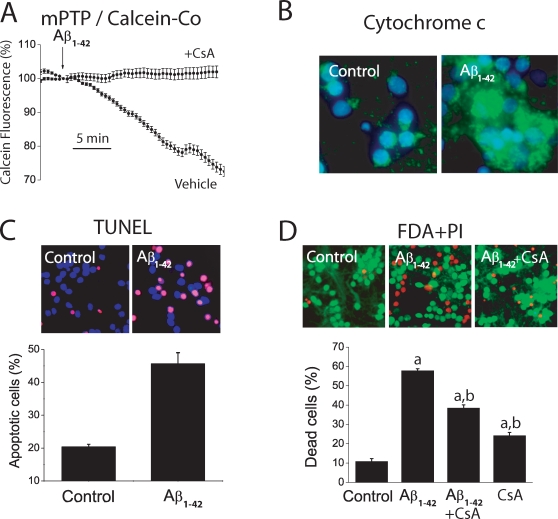
References
-
- Wegiel J, Wisniewski HM, Dziewiatkowski J, Badmajew E, Tarnawski M, et al. Cerebellar atrophy in Alzheimer's disease-clinicopathological correlations. Brain Res. 1999;818:41–50. - PubMed
-
- Allen JW, Eldadah BA, Faden AI. Beta-amyloid-induced apoptosis of cerebellar granule cells and cortical neurons: exacerbation by selective inhibition of group I metabotropic glutamate receptors. Neuropharmacology. 1999;38:1243–1252. - PubMed
-
- Canu N, Calissano P. In vitro cultured neurons for molecular studies correlating apoptosis with events related to Alzheimer disease. Cerebellum. 2003;2:270–278. - PubMed
-
- Plant LD, Webster NJ, Boyle JP, Ramsden M, Freir DB, et al. Amyloid beta peptide as a physiological modulator of neuronal ‘A’-type K+ current. Neurobiol Aging. 2006;11:1673–1683. - PubMed
-
- McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
