

Molecular subsets in the gene expression signatures of scleroderma skin
- PMID: 18648520
- PMCID: PMC2481301
- DOI: 10.1371/journal.pone.0002696
Molecular subsets in the gene expression signatures of scleroderma skin
Erratum in
- PLoS ONE. 2008;3(10). doi: 10.1371/annotation/05bed72c-c6f6-4685-a732-02c78e5f66c2 doi: 10.1371/annotation/05bed72c-c6f6-4685-a732-02c78e5f66c2
Abstract
Background: Scleroderma is a clinically heterogeneous disease with a complex phenotype. The disease is characterized by vascular dysfunction, tissue fibrosis, internal organ dysfunction, and immune dysfunction resulting in autoantibody production.
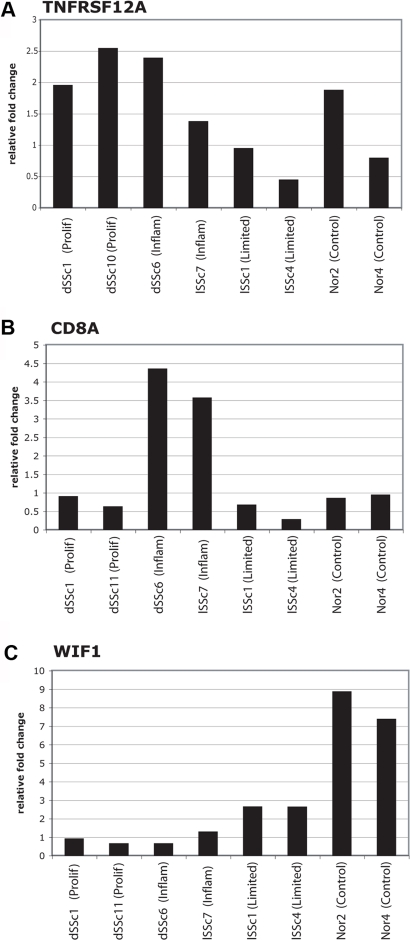
Methodology and findings: We analyzed the genome-wide patterns of gene expression with DNA microarrays in skin biopsies from distinct scleroderma subsets including 17 patients with systemic sclerosis (SSc) with diffuse scleroderma (dSSc), 7 patients with SSc with limited scleroderma (lSSc), 3 patients with morphea, and 6 healthy controls. 61 skin biopsies were analyzed in a total of 75 microarray hybridizations. Analysis by hierarchical clustering demonstrates nearly identical patterns of gene expression in 17 out of 22 of the forearm and back skin pairs of SSc patients. Using this property of the gene expression, we selected a set of 'intrinsic' genes and analyzed the inherent data-driven groupings. Distinct patterns of gene expression separate patients with dSSc from those with lSSc and both are easily distinguished from normal controls. Our data show three distinct patient groups among the patients with dSSc and two groups among patients with lSSc. Each group can be distinguished by unique gene expression signatures indicative of proliferating cells, immune infiltrates and a fibrotic program. The intrinsic groups are statistically significant (p<0.001) and each has been mapped to clinical covariates of modified Rodnan skin score, interstitial lung disease, gastrointestinal involvement, digital ulcers, Raynaud's phenomenon and disease duration. We report a 177-gene signature that is associated with severity of skin disease in dSSc.
Conclusions and significance: Genome-wide gene expression profiling of skin biopsies demonstrates that the heterogeneity in scleroderma can be measured quantitatively with DNA microarrays. The diversity in gene expression demonstrates multiple distinct gene expression programs in the skin of patients with scleroderma.
Conflict of interest statement
Figures






References
-
- Mayes MD. Classification and epidemiology of scleroderma. Semin Cutan Med Surg. 1998;17:22–26. - PubMed
-
- Mayes MD, Lacey JV, Jr., Beebe-Dimmer J, Gillespie BW, Cooper B, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246–2255. - PubMed
-
- Leroy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. JRheumatol. 1988;15:202–205. - PubMed
-
- Medsger TA. Systemic sclerosis (scleroderma): clinical aspects. In: Koopman WJ, editor. Arthritis and Allied Conditions. 14th ed. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 1590.
-
- Masi AT. Classification of systemic sclerosis (scleroderma): relationship of cutaneous subgroups in early disease to outcome and serologic reactivity. J Rheumatol. 1988;15:894–898. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
