

A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression
- PMID: 18650261
- PMCID: PMC2544442
- DOI: 10.1152/ajpcell.00554.2007
A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression
Abstract
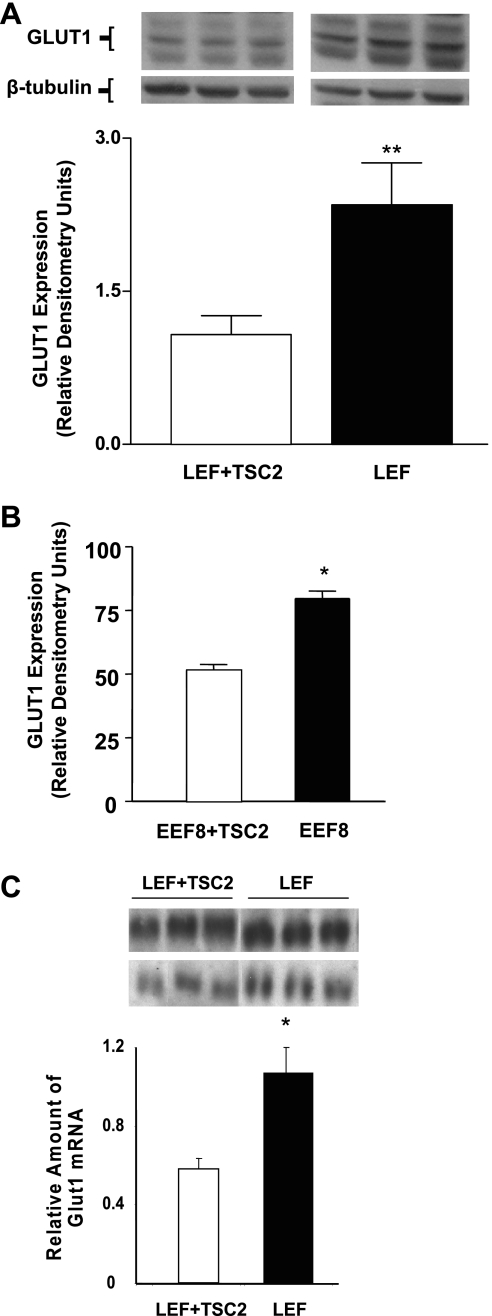
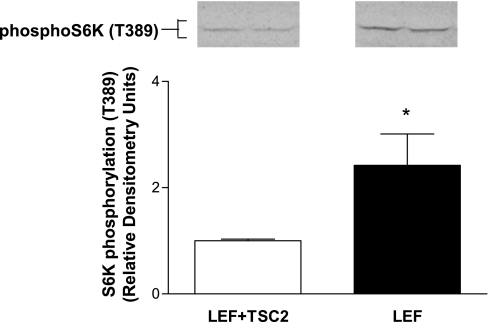
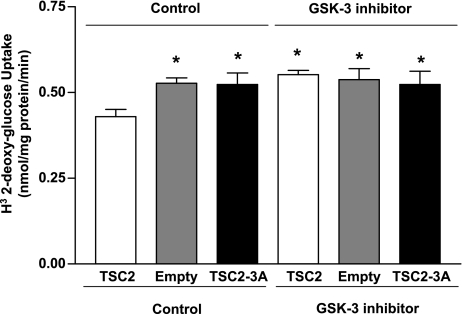
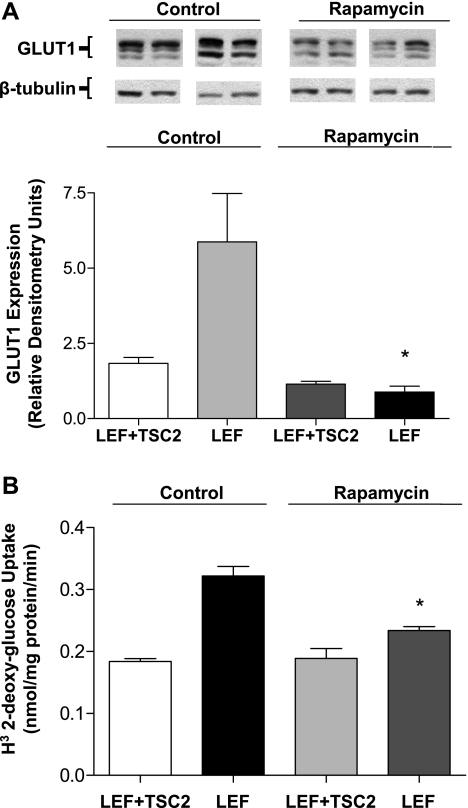
Glucose transport is a highly regulated process and is dependent on a variety of signaling events. Glycogen synthase kinase-3 (GSK-3) has been implicated in various aspects of the regulation of glucose transport, but the mechanisms by which GSK-3 activity affects glucose uptake have not been well defined. We report that basal glycogen synthase kinase-3 (GSK-3) activity regulates glucose transport in several cell types. Chronic inhibition of basal GSK-3 activity (8-24 h) in several cell types, including vascular smooth muscle cells, resulted in an approximately twofold increase in glucose uptake due to a similar increase in protein expression of the facilitative glucose transporter 1 (GLUT1). Conversely, expression of a constitutively active form of GSK-3beta resulted in at least a twofold decrease in GLUT1 expression and glucose uptake. Since GSK-3 can inhibit mammalian target of rapamycin (mTOR) signaling via phosphorylation of the tuberous sclerosis complex subunit 2 (TSC2) tumor suppressor, we investigated whether chronic GSK-3 effects on glucose uptake and GLUT1 expression depended on TSC2 phosphorylation and TSC inhibition of mTOR. We found that absence of functional TSC2 resulted in a 1.5-to 3-fold increase in glucose uptake and GLUT1 expression in multiple cell types. These increases in glucose uptake and GLUT1 levels were prevented by inhibition of mTOR with rapamycin. GSK-3 inhibition had no effect on glucose uptake or GLUT1 expression in TSC2 mutant cells, indicating that GSK-3 effects on GLUT1 and glucose uptake were mediated by a TSC2/mTOR-dependent pathway. The effect of GSK-3 inhibition on GLUT1 expression and glucose uptake was restored in TSC2 mutant cells by transfection of a wild-type TSC2 vector, but not by a TSC2 construct with mutated GSK-3 phosphorylation sites. Thus, TSC2 and rapamycin-sensitive mTOR function downstream of GSK-3 to modulate effects of GSK-3 on glucose uptake and GLUT1 expression. GSK-3 therefore suppresses glucose uptake via TSC2 and mTOR and may serve to match energy substrate utilization to cellular growth.
Figures





References
-
- Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol Res 50: 545–549, 2004. - PubMed
-
- Atkins KB, Johns D, Watts S, Webb CR, Brosius FC. Decreased vascular glucose transporter expression and glucose uptake in DOCA-salt hypertension. J Hypertens 19: 1581–1587, 2001. - PubMed
-
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer L, Foufelle F, Carling D, Hardie DG, Baldwin SA. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 115: 2433–2442, 2002. - PubMed
-
- Bentley J, Itchayanan D, Barnes K, McIntosh E, Tang X, Downes CP, Holman GD, Whetton AD, Owen-Lynch PJ, Baldwin SA. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J Biol Chem 278: 39337–39348, 2003. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous