

In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia
- PMID: 18650378
- PMCID: PMC2492493
- DOI: 10.1073/pnas.0711666105
In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia
Abstract
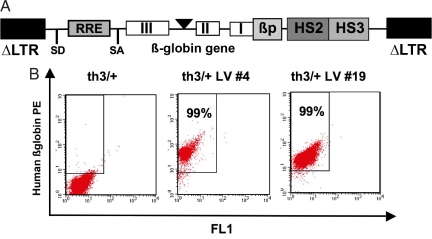
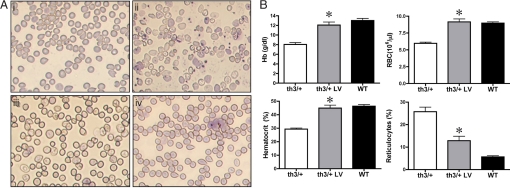
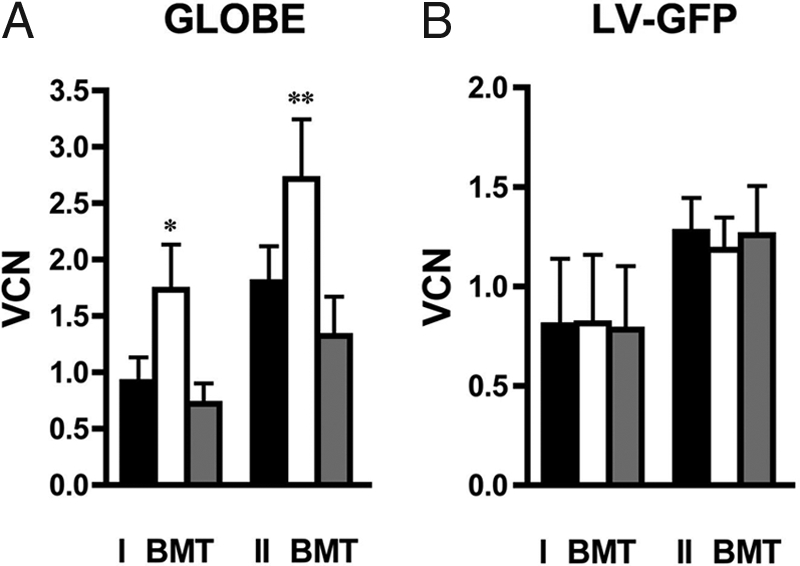
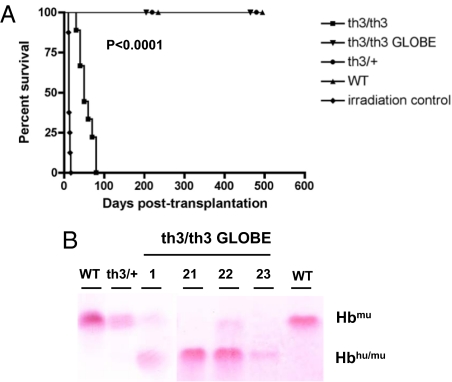
Gene therapy for beta-thalassemia requires stable transfer of a beta-globin gene into hematopoietic stem cells (HSCs) and high and regulated hemoglobin expression in the erythroblastic progeny. We developed an erythroid-specific lentiviral vector driving the expression of the human beta-globin gene from a minimal promoter/enhancer element containing two hypersensitive sites from the beta-globin locus control region. Transplantation of transduced HSCs into thalassemic mice leads to stable and long-term correction of anemia with all red blood cells expressing the transgene. A frequency of 30-50% of transduced HSCs, harboring an average vector copy number per cell of 1, was sufficient to fully correct the thalassemic phenotype. In the mouse model of Cooley's anemia transplantation of transduced cells rescues lethality, leading to either a normal or a thalassemia intermedia phenotype. We show that genetically corrected erythroblasts undergo in vivo selection with preferential survival of progenitors harboring proviral integrations in genome sites more favorable to high levels of vector-derived expression. These data provide a rationale for a gene therapy approach to beta-thalassemia based on partially myeloablative transplantation protocols.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lucarelli G, Andreani M, Angelucci E. The cure of thalassemia by bone marrow transplantation. Blood Rev. 2002;16:81–85. - PubMed
-
- May C, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. - PubMed
-
- Puthenveetil G, et al. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. - PubMed
-
- Sadelain M. Recent advances in globin gene transfer for the treatment of beta-thalassemia and sickle cell anemia. Curr Opin Hematol. 2006;13:142–148. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
