

From endoplasmic-reticulum stress to the inflammatory response
- PMID: 18650916
- PMCID: PMC2727659
- DOI: 10.1038/nature07203
From endoplasmic-reticulum stress to the inflammatory response
Abstract
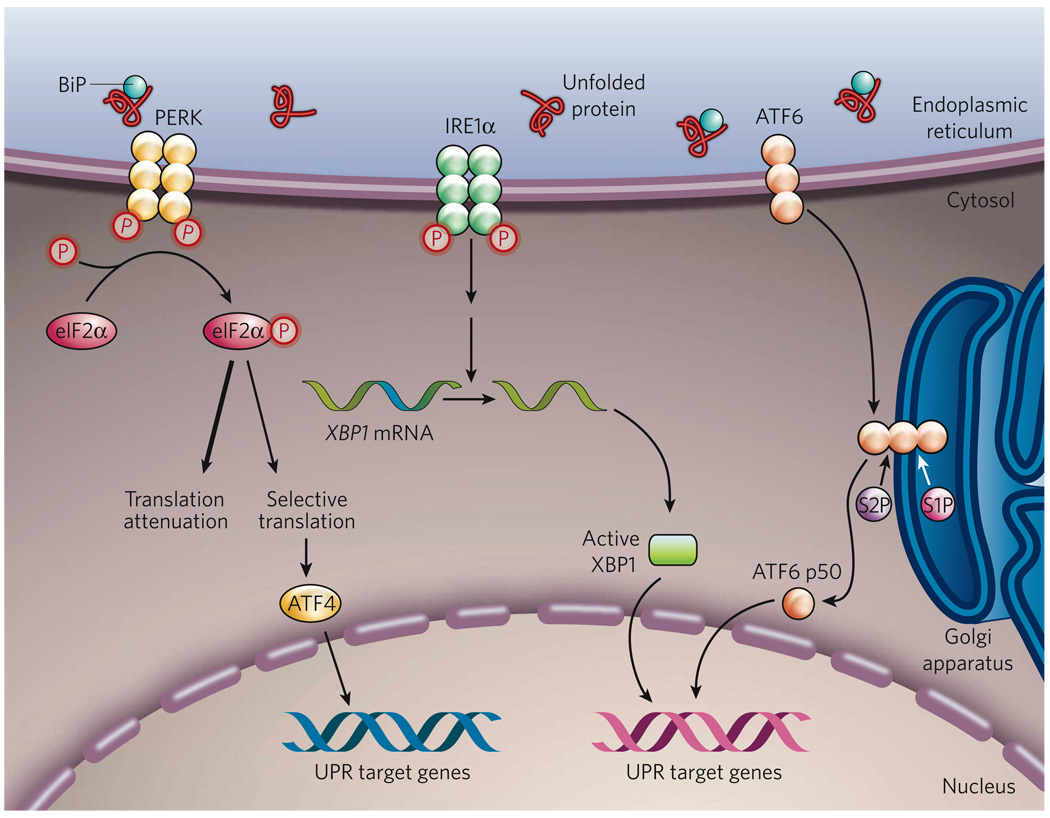
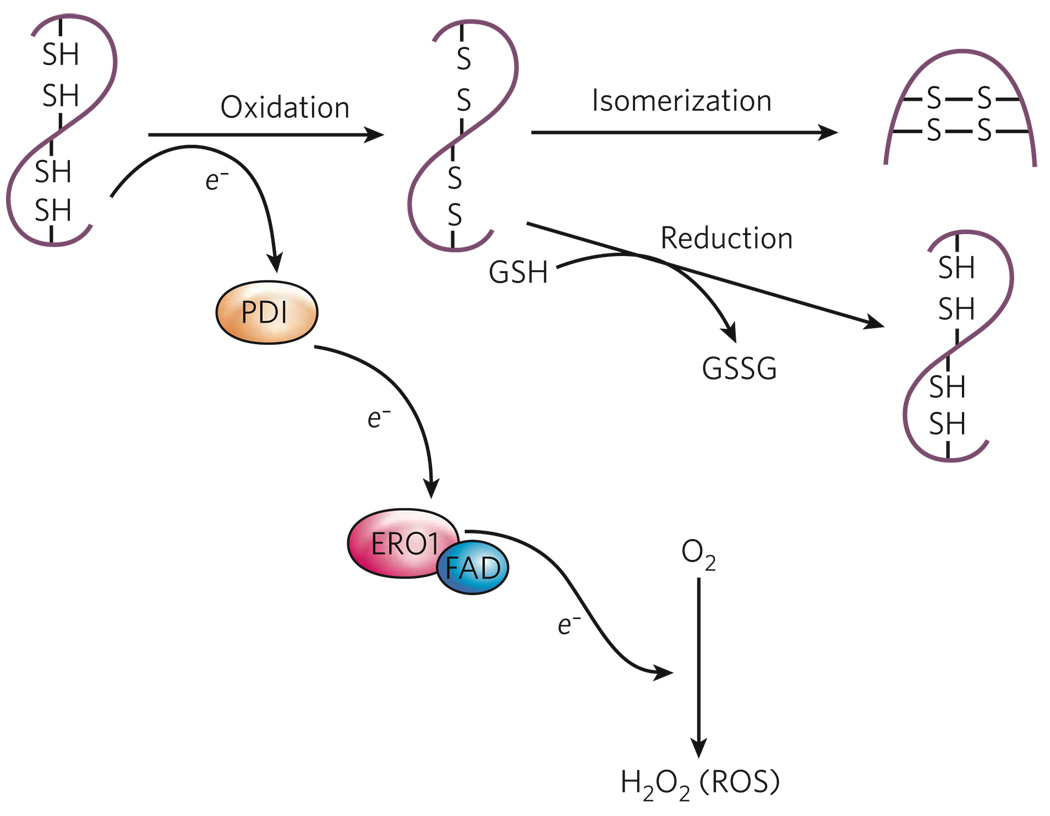
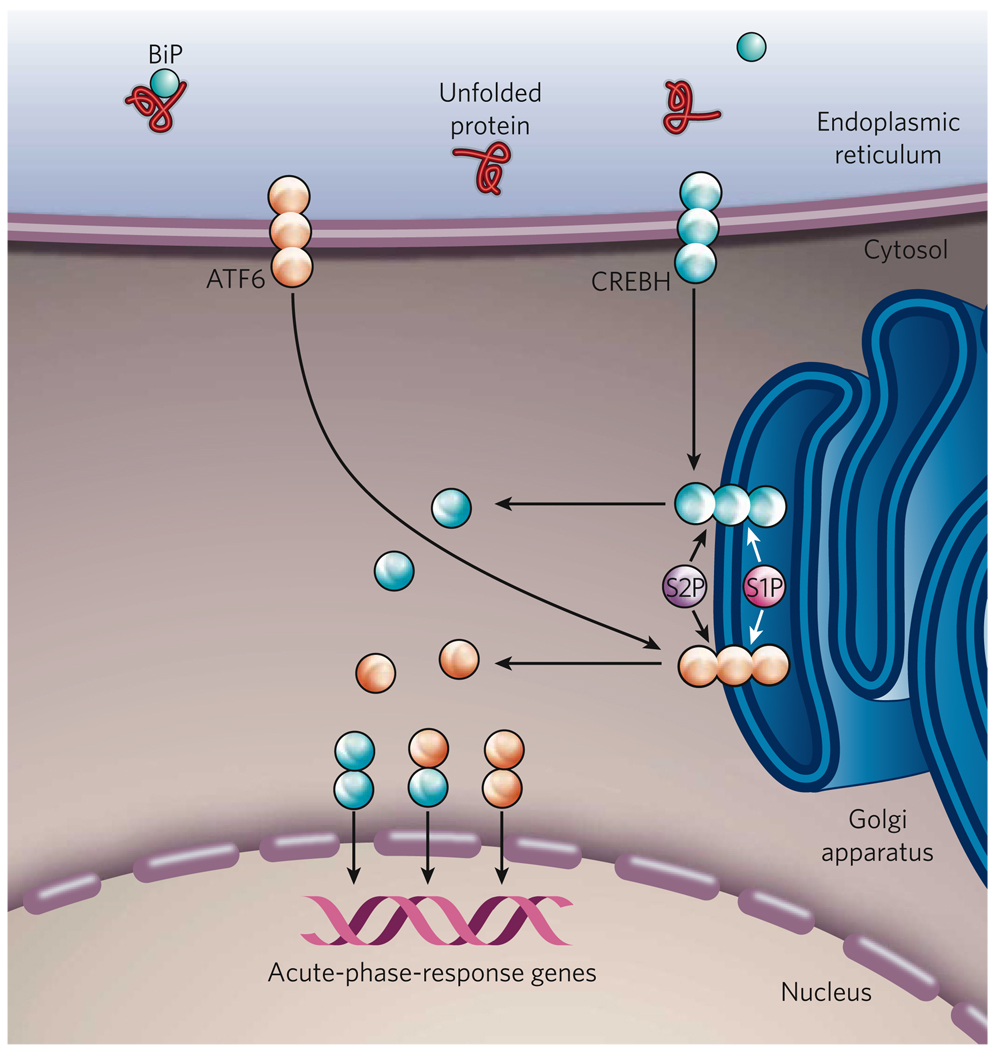
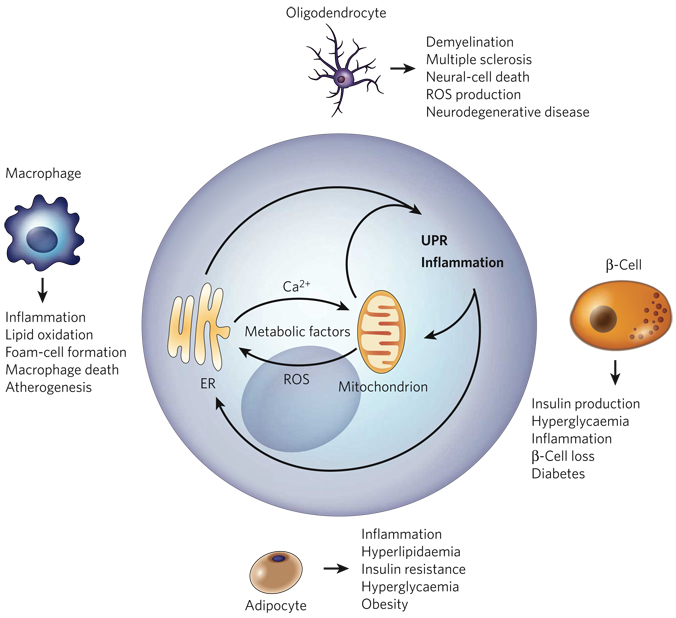
The endoplasmic reticulum is responsible for much of a cell's protein synthesis and folding, but it also has an important role in sensing cellular stress. Recently, it has been shown that the endoplasmic reticulum mediates a specific set of intracellular signalling pathways in response to the accumulation of unfolded or misfolded proteins, and these pathways are collectively known as the unfolded-protein response. New observations suggest that the unfolded-protein response can initiate inflammation, and the coupling of these responses in specialized cells and tissues is now thought to be fundamental in the pathogenesis of inflammatory diseases. The knowledge gained from this emerging field will aid in the development of therapies for modulating cellular stress and inflammation.
Figures


References
-
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006;354:610–621. - PubMed
-
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. - PubMed
-
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nature Rev. Immunol. 2006;6:508–519. - PubMed
-
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. - PubMed
-
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Rev. Mol. Cell Biol. 2007;8:519–529. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
