

Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones
- PMID: 18650938
- PMCID: PMC2516887
- DOI: 10.1038/emboj.2008.139
Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones
Abstract
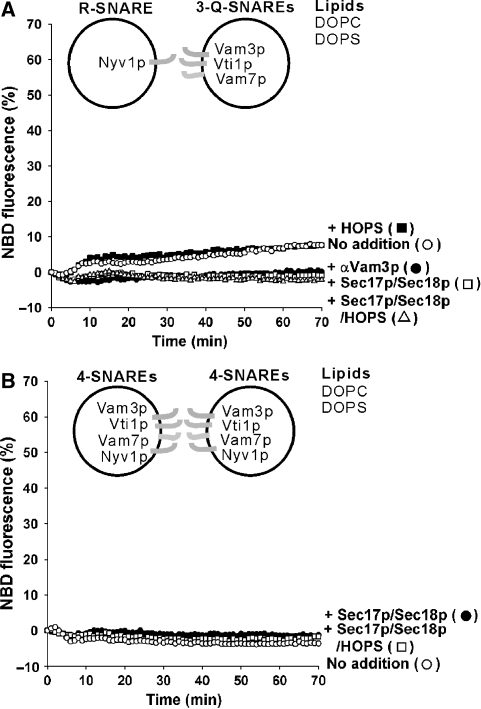
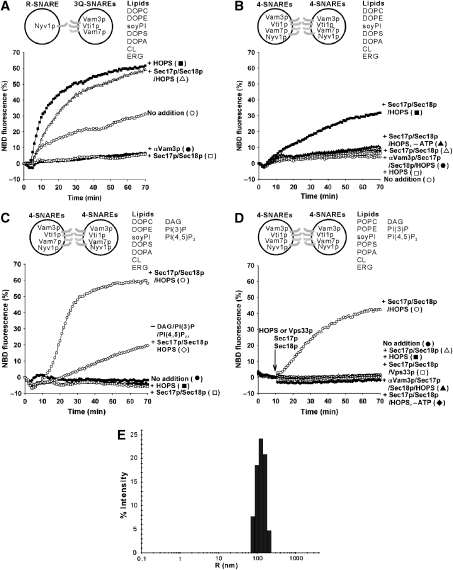
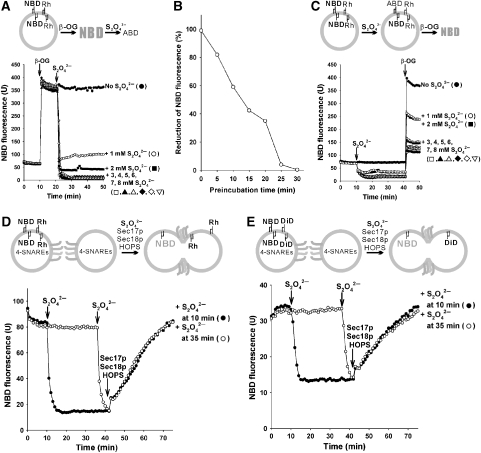
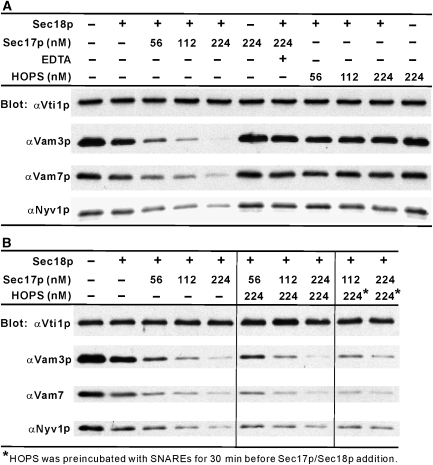
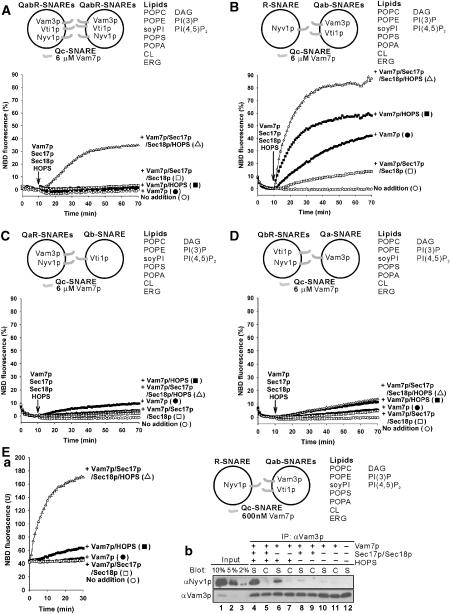
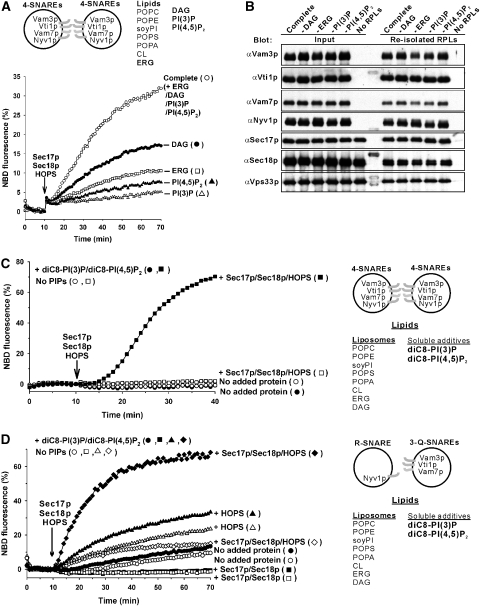
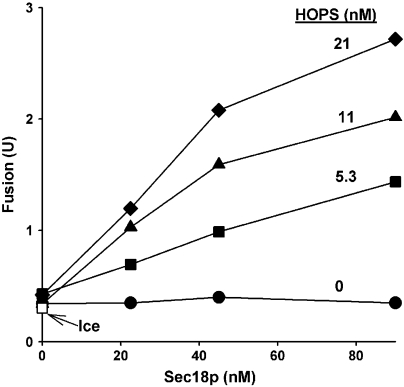
The homotypic fusion of yeast vacuoles, each with 3Q- and 1R-SNARE, requires SNARE chaperones (Sec17p/Sec18p and HOPS) and regulatory lipids (sterol, diacylglycerol and phosphoinositides). Pairs of liposomes of phosphatidylcholine/phosphatidylserine, bearing three vacuolar Q-SNAREs on one and the R-SNARE on the other, undergo slow lipid mixing, but this is unaffected by HOPS and inhibited by Sec17p/Sec18p. To study these essential fusion components, we reconstituted proteoliposomes of a more physiological composition, bearing vacuolar lipids and all four vacuolar SNAREs. Their fusion requires Sec17p/Sec18p and HOPS, and each regulatory lipid is important for rapid fusion. Although SNAREs can cause both fusion and lysis, fusion of these proteoliposomes with Sec17p/Sec18p and HOPS is not accompanied by lysis. Sec17p/Sec18p, which disassemble SNARE complexes, and HOPS, which promotes and proofreads SNARE assembly, act synergistically to form fusion-competent SNARE complexes, and this synergy requires phosphoinositides. This is the first chemically defined model of the physiological interactions of these conserved fusion catalysts.
Figures
References
-
- Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Südhof TC, Rizo J (2006) Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol 13: 209–217 - PubMed
-
- Bhalla A, Chicka MC, Tucker WC, Chapman ER (2006) Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol 13: 323–330 - PubMed
-
- Burgess SW, McIntosh TJ, Lentz BR (1992) Modulation of Poly (ethylene glycol)-induced fusion by membrane hydration: importance of interbilayer separation. Biochemistry 31: 2653–2661 - PubMed
-
- Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol 3: 613–618 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
