

Dynamics of genome rearrangement in bacterial populations
- PMID: 18650965
- PMCID: PMC2483231
- DOI: 10.1371/journal.pgen.1000128
Dynamics of genome rearrangement in bacterial populations
Abstract
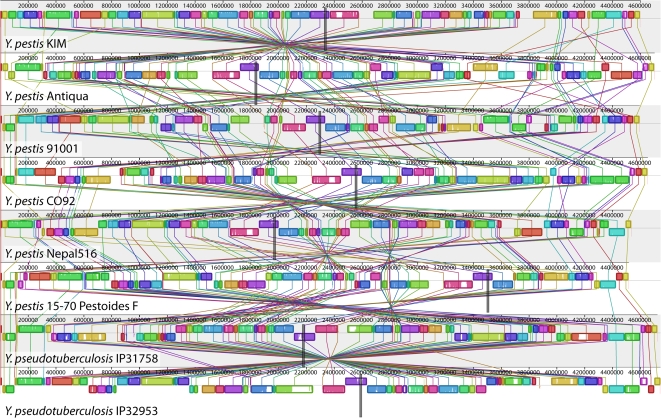
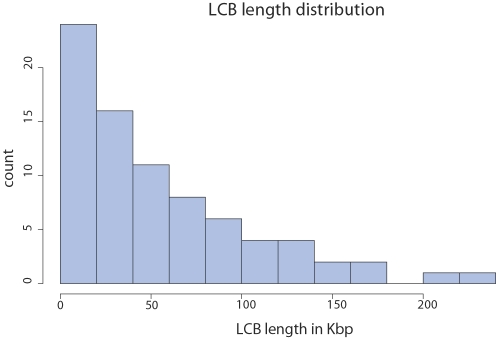
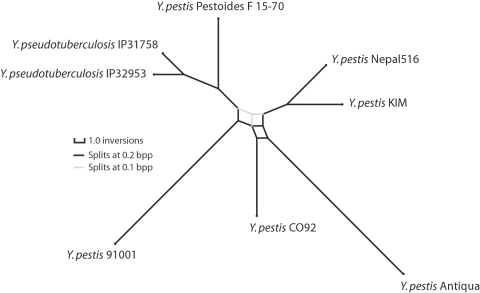
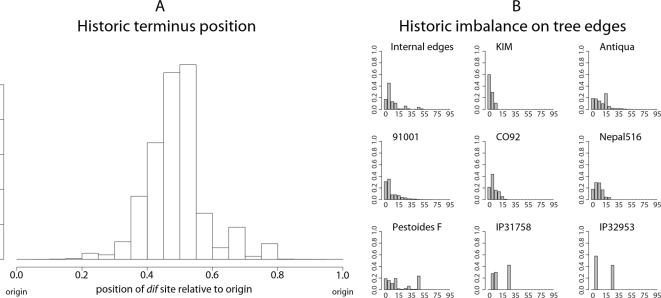
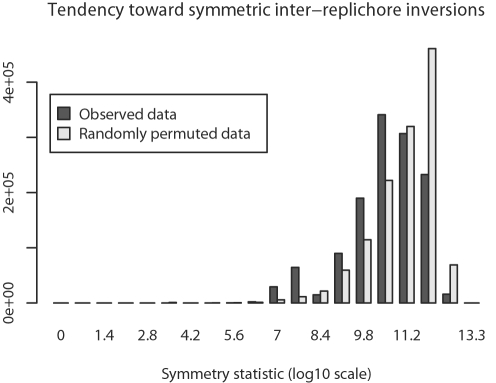
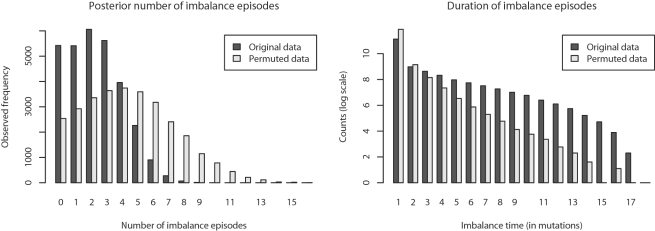
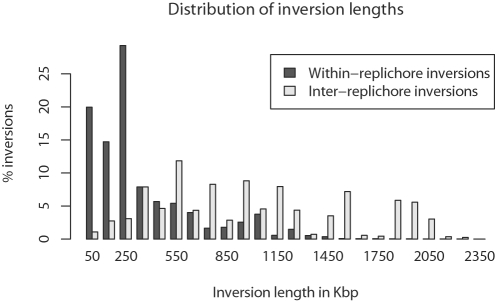
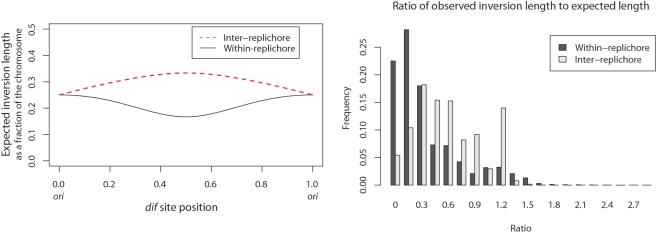
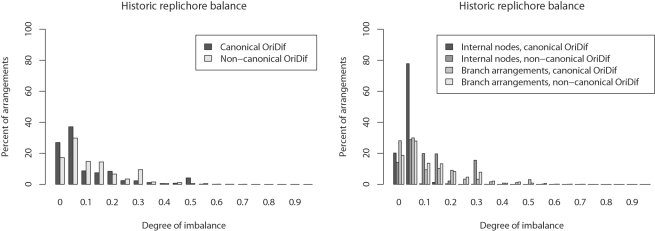
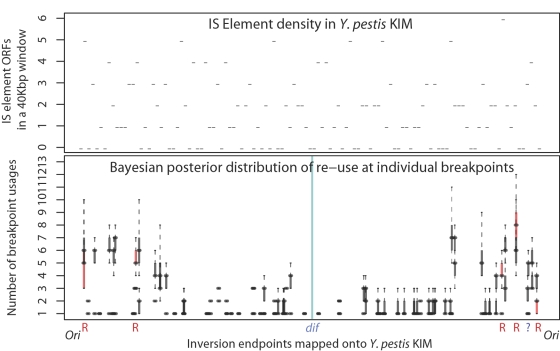
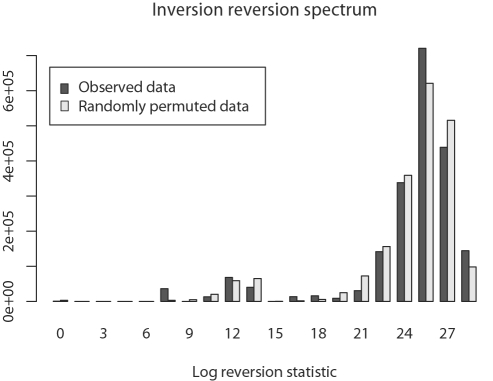
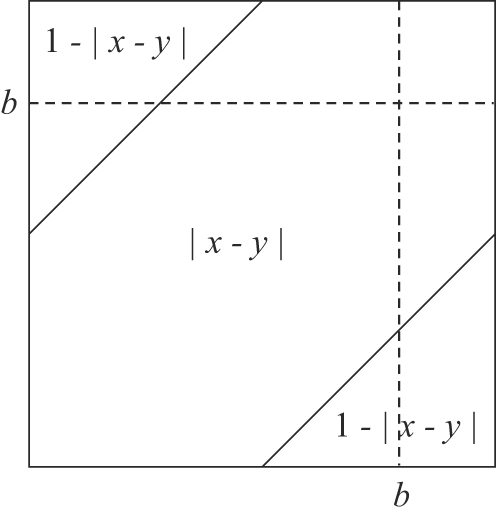
Genome structure variation has profound impacts on phenotype in organisms ranging from microbes to humans, yet little is known about how natural selection acts on genome arrangement. Pathogenic bacteria such as Yersinia pestis, which causes bubonic and pneumonic plague, often exhibit a high degree of genomic rearrangement. The recent availability of several Yersinia genomes offers an unprecedented opportunity to study the evolution of genome structure and arrangement. We introduce a set of statistical methods to study patterns of rearrangement in circular chromosomes and apply them to the Yersinia. We constructed a multiple alignment of eight Yersinia genomes using Mauve software to identify 78 conserved segments that are internally free from genome rearrangement. Based on the alignment, we applied Bayesian statistical methods to infer the phylogenetic inversion history of Yersinia. The sampling of genome arrangement reconstructions contains seven parsimonious tree topologies, each having different histories of 79 inversions. Topologies with a greater number of inversions also exist, but were sampled less frequently. The inversion phylogenies agree with results suggested by SNP patterns. We then analyzed reconstructed inversion histories to identify patterns of rearrangement. We confirm an over-representation of "symmetric inversions"-inversions with endpoints that are equally distant from the origin of chromosomal replication. Ancestral genome arrangements demonstrate moderate preference for replichore balance in Yersinia. We found that all inversions are shorter than expected under a neutral model, whereas inversions acting within a single replichore are much shorter than expected. We also found evidence for a canonical configuration of the origin and terminus of replication. Finally, breakpoint reuse analysis reveals that inversions with endpoints proximal to the origin of DNA replication are nearly three times more frequent. Our findings represent the first characterization of genome arrangement evolution in a bacterial population evolving outside laboratory conditions. Insight into the process of genomic rearrangement may further the understanding of pathogen population dynamics and selection on the architecture of circular bacterial chromosomes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Sousa C, de Lorenzo V, Cebolla A. Modulation of gene expression through chromosomal positioning in Escherichia coli. Microbiology. 1997;143 (Pt 6):2071–2078. - PubMed
-
- Couturier E, Rocha EP. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Molecular Microbiology. 2006;59:1506–1518. doi:10.1111/j.1365-2958.2006.05046.x. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
