

Population genomic analysis of strain variation in Leptospirillum group II bacteria involved in acid mine drainage formation
- PMID: 18651792
- PMCID: PMC2475542
- DOI: 10.1371/journal.pbio.0060177
Population genomic analysis of strain variation in Leptospirillum group II bacteria involved in acid mine drainage formation
Abstract
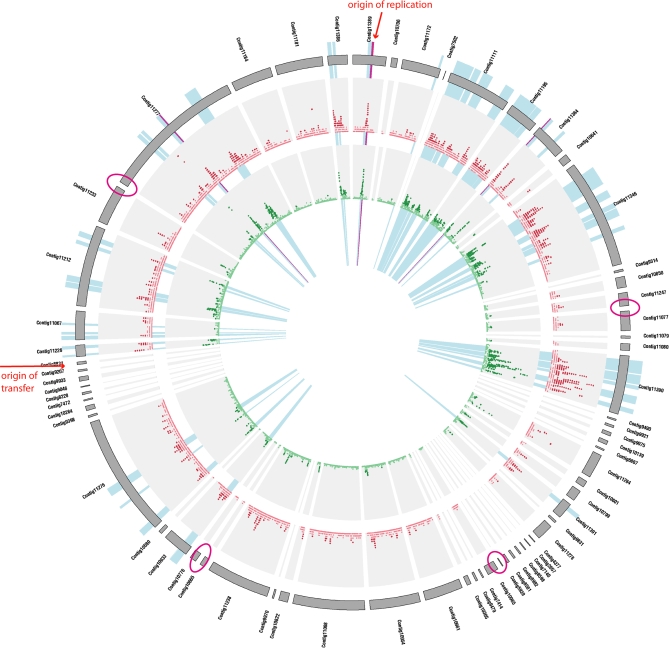
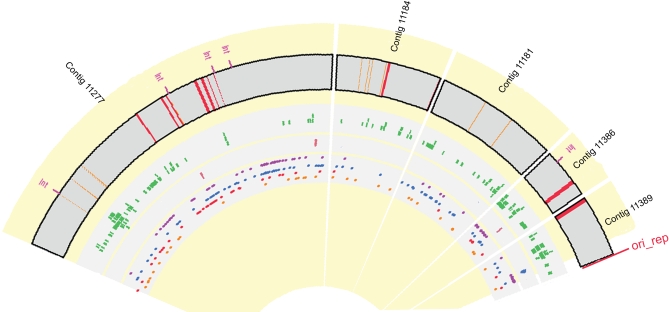
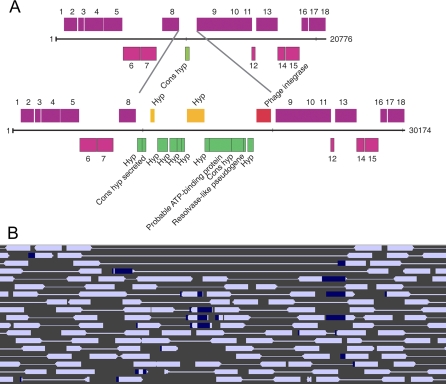
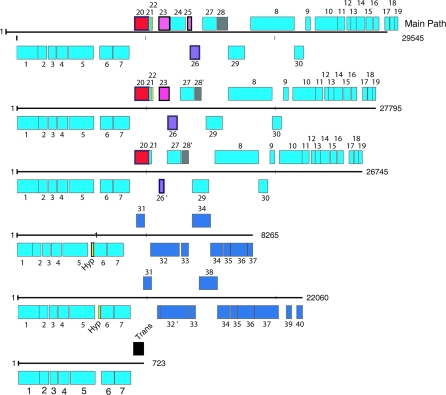
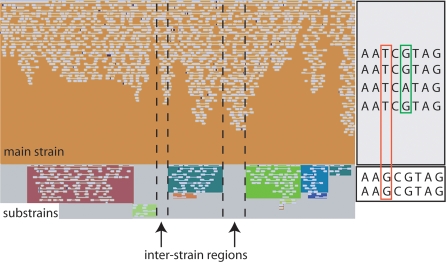
Deeply sampled community genomic (metagenomic) datasets enable comprehensive analysis of heterogeneity in natural microbial populations. In this study, we used sequence data obtained from the dominant member of a low-diversity natural chemoautotrophic microbial community to determine how coexisting closely related individuals differ from each other in terms of gene sequence and gene content, and to uncover evidence of evolutionary processes that occur over short timescales. DNA sequence obtained from an acid mine drainage biofilm was reconstructed, taking into account the effects of strain variation, to generate a nearly complete genome tiling path for a Leptospirillum group II species closely related to L. ferriphilum (sampling depth approximately 20x). The population is dominated by one sequence type, yet we detected evidence for relatively abundant variants (>99.5% sequence identity to the dominant type) at multiple loci, and a few rare variants. Blocks of other Leptospirillum group II types ( approximately 94% sequence identity) have recombined into one or more variants. Variant blocks of both types are more numerous near the origin of replication. Heterogeneity in genetic potential within the population arises from localized variation in gene content, typically focused in integrated plasmid/phage-like regions. Some laterally transferred gene blocks encode physiologically important genes, including quorum-sensing genes of the LuxIR system. Overall, results suggest inter- and intrapopulation genetic exchange involving distinct parental genome types and implicate gain and loss of phage and plasmid genes in recent evolution of this Leptospirillum group II population. Population genetic analyses of single nucleotide polymorphisms indicate variation between closely related strains is not maintained by positive selection, suggesting that these regions do not represent adaptive differences between strains. Thus, the most likely explanation for the observed patterns of polymorphism is divergence of ancestral strains due to geographic isolation, followed by mixing and subsequent recombination.
Conflict of interest statement
Figures


References
-
- Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, et al. Genomic islands and the ecology and evolution of Prochlorococcus . Science. 2006;311:1768–1770. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
