

Alpha-mannosidosis
- PMID: 18651971
- PMCID: PMC2515294
- DOI: 10.1186/1750-1172-3-21
Alpha-mannosidosis
Abstract
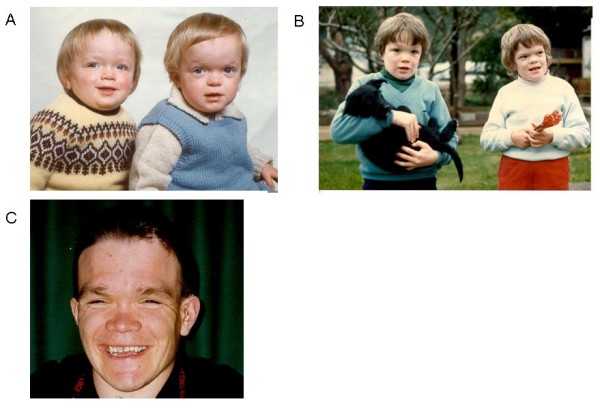
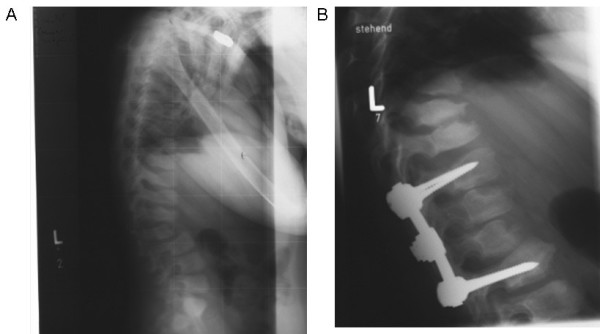
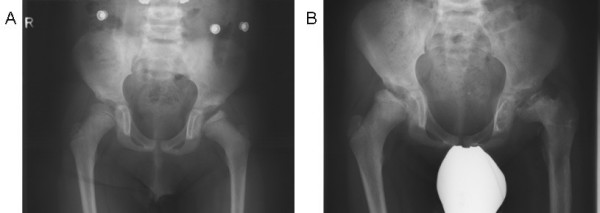
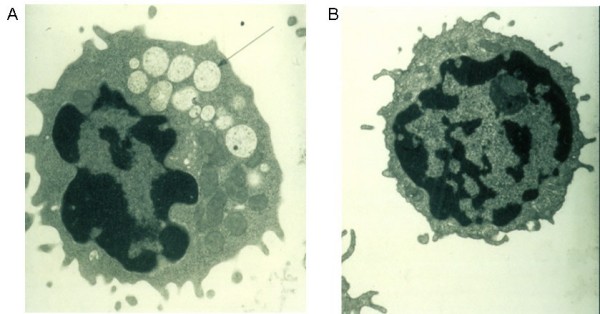
Alpha-mannosidosis is an inherited lysosomal storage disorder characterized by immune deficiency, facial and skeletal abnormalities, hearing impairment, and intellectual disability. It occurs in approximately 1 of 500,000 live births. The children are often born apparently normal, and their condition worsens progressively. Some children are born with ankle equinus or develop hydrocephalus in the first year of life. Main features are immune deficiency (manifested by recurrent infections, especially in the first decade of life), skeletal abnormalities (mild-to-moderate dysostosis multiplex, scoliosis and deformation of the sternum), hearing impairment (moderate-to-severe sensorineural hearing loss), gradual impairment of mental functions and speech, and often, periods of psychosis. Associated motor function disturbances include muscular weakness, joint abnormalities and ataxia. The facial trait include large head with prominent forehead, rounded eyebrows, flattened nasal bridge, macroglossia, widely spaced teeth, and prognathism. Slight strabismus is common. The clinical variability is significant, representing a continuum in severity. The disorder is caused by lysosomal alpha-mannosidase deficiency. Alpha-mannosidosis is inherited in an autosomal recessive fashion and is caused by mutations in the MAN2B1 gene located on chromosome 19 (19 p13.2-q12). Diagnosis is made by measuring acid alpha-mannosidase activity in leukocytes or other nucleated cells and can be confirmed by genetic testing. Elevated urinary secretion of mannose-rich oligosaccharides is suggestive, but not diagnostic. Differential diagnoses are mainly the other lysosomal storage diseases like the mucopolysaccharidoses. Genetic counseling should be given to explain the nature of the disease and to detect carriers. Antenatal diagnosis is possible, based on both biochemical and genetic methods. The management should be pro-active, preventing complications and treating manifestations. Infections must be treated frequently. Otolaryngological treatment of fluid in the middle ear is often required and use of hearing aids is invariably required. Early educational intervention for development of social skills is needed and physiotherapy is important to improve bodily function. Orthopedic surgery may be necessary. The long-term prognosis is poor. There is an insidiously slow progression of neuromuscular and skeletal deterioration over several decades, making most patients wheel-chair dependent. No patients manage to be completely socially independent. Many patients are over 50 years of age.
Figures
References
-
- Ockerman PA. A generalised storage disorder resembling Hurler's syndrome. Lancet. 1967;2:239. doi: 10.1016/S0140-6736(67)92303-3. - DOI
-
- Loeb H, Vamos-Hurwitz E. Mannosidosis. Arch Neurol. 1977;34:650–651. - PubMed
-
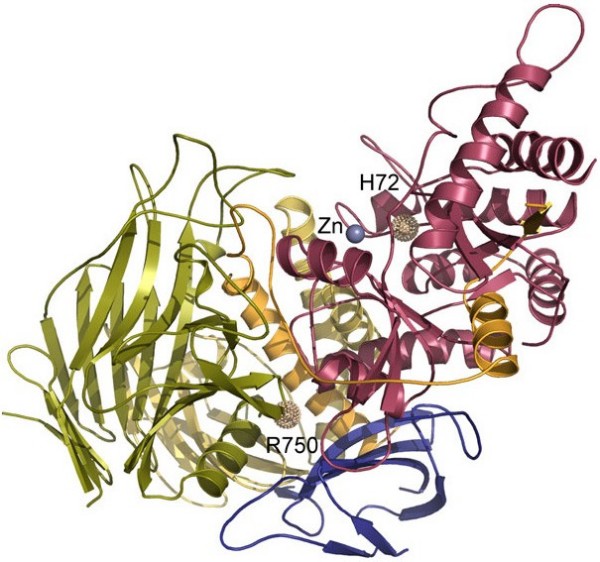
- Heikinheimo P, Helland R, Leiros HK, Leiros I, Karlsen S, Evjen G, Ravelli R, Schoehn G, Ruigrok R, Tollersrud OK, McSweeney S, Hough E. The structure of bovine lysosomal alpha-mannosidase suggests a novel mechanism for low-pH activation. J Mol Biol. 2003;327:631–644. doi: 10.1016/S0022-2836(03)00172-4. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
