

Review
doi: 10.1038/nri2361.
Genome-wide association studies: a new window into immune-mediated diseases
Affiliations
- PMID: 18654571
- PMCID: PMC2750781
- DOI: 10.1038/nri2361
Item in Clipboard
Review
Genome-wide association studies: a new window into immune-mediated diseases
Nat Rev Immunol.
2008 Aug.
Abstract
Given the recent explosion of genetic discoveries, 2007 is becoming known to human geneticists as the year of genome-wide association studies. In fact, more genetic risk factors for common diseases were identified in this one year than had been collectively reported before 2007. In particular, 2007 witnessed the discovery of many genes that influence susceptibility to individual immune-mediated diseases, as well as other genes that are associated with susceptibility to more than one disease. Although much work remains to be done, in this Review we discuss what effect these studies are having on our understanding of disease pathogenesis and their potential impact on future immunology studies.
Figures
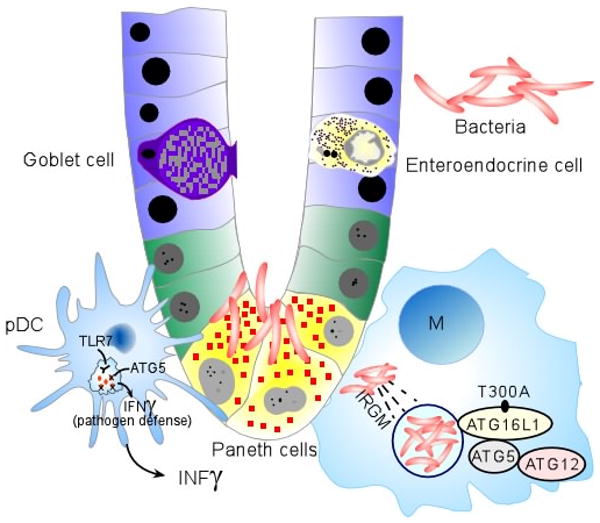
(a) Under homeostatic conditions, the mucus layer and tight junctions associated with intestinal epithelial cells maintain barrier integrity. A dynamic balance between host defence immune responses and luminal enteric bacteria at the mucosal frontier is central to the pathogenesis of Crohn's disease. Recent genetics studies have implicated autophagy, defects in innate immunity and imbalance between pathogenic and regulatory T cell populations as risk factors for Crohn's disease. Dendritic cells (DCs) extend dendrites between epithelial cells and can sense changes in luminal contents. Potential signalling pathways in inflammation, and the respective genes involved, are represented. Caspase-recruitment domain 15 (NOD2) has a role in the release of antimicrobial peptides and in sensing cytosolic microbial ligands; prostaglandin E receptor 4 (PTGER4) is involved in promoting epithelial restitution; the interleukin-23 receptor (IL23R) has a role in generating and maintaining of TH17 cells; autophagy 16-like 1 (ATG16L1) and immunity-related GTPase family member M (IRGM) are involved in and microbial defence and adaptive immunity; macrophage-stimulating factor 1 (MST1) has a role in down regulating inflammatory response.
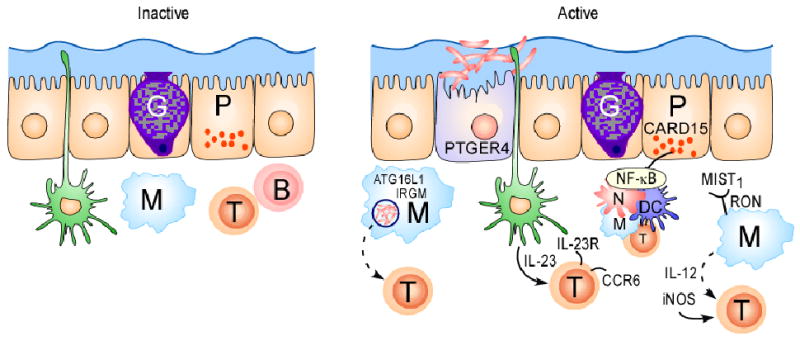
(a) Under homeostatic conditions, the mucus layer and tight junctions associated with intestinal epithelial cells maintain barrier integrity. A dynamic balance between host defence immune responses and luminal enteric bacteria at the mucosal frontier is central to the pathogenesis of Crohn's disease. Recent genetics studies have implicated autophagy, defects in innate immunity and imbalance between pathogenic and regulatory T cell populations as risk factors for Crohn's disease. Dendritic cells (DCs) extend dendrites between epithelial cells and can sense changes in luminal contents. Potential signalling pathways in inflammation, and the respective genes involved, are represented. Caspase-recruitment domain 15 (NOD2) has a role in the release of antimicrobial peptides and in sensing cytosolic microbial ligands; prostaglandin E receptor 4 (PTGER4) is involved in promoting epithelial restitution; the interleukin-23 receptor (IL23R) has a role in generating and maintaining of TH17 cells; autophagy 16-like 1 (ATG16L1) and immunity-related GTPase family member M (IRGM) are involved in and microbial defence and adaptive immunity; macrophage-stimulating factor 1 (MST1) has a role in down regulating inflammatory response.
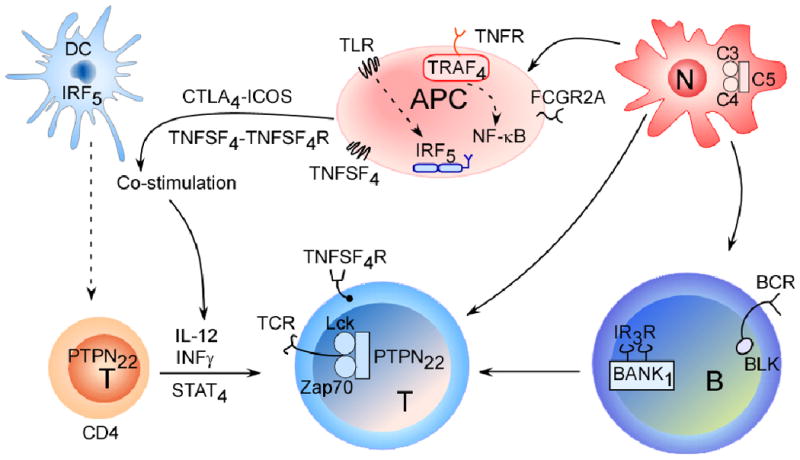
GWAS have highlighted both common and unique genetic variants that are associated with rheumatoid arthritis and systemic lupus erythematosus (SLE). The depicted cells and signalling pathways illustrate the multiple components of innate and adaptive immune responses that can contribute to autoimmune responses in these diseases. The proteins encoded by candidate genes associated with rheumatoid arthritis and SLE are depicted. Ultimately collaboration between different cell types initiates local and systemic inflammation. The complement cascade (involving complement component 5 (C5)), which influences both rheumatoid arthritis and SLE, reports danger signals to the surrounding mileu through neutrophil recruitment and activation. Antigen presenting cells are equipped to eliminate pathogens and provide cytokines and co-stimulatory cues (such as tumour-necrosis factor superfamily 4 (TNFSF4)) to T cells. Other adaptor proteins, such as tumour-necrosis factor receptor-associated factor 4 (TRAF4), and the activation of transcription factors such as signal transducer and activator of transcription 4 (STAT4) and interferon-regulatory factor 5 (IRF5), which is involved in type 1 interferon (IFN) production, direct the secretion of specific cytokines and chemokines. The involvement of autoantibodies (which are cell-derived) as well as type 1 IFNs in classic SLE demonstrate that disease pathology involves a dysfunction in both the innate and adaptive immune responses.
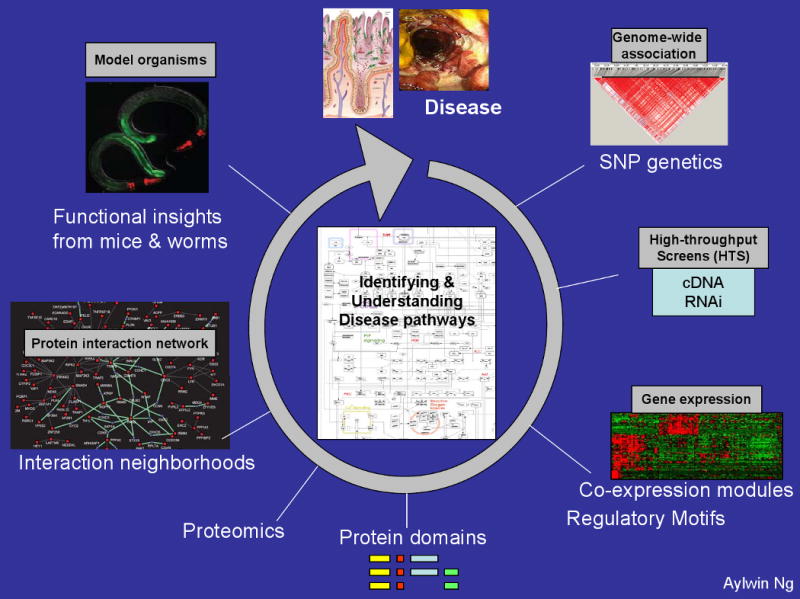
a) Fine mapping of causal variants associated with disease susceptibility. Genes identified as associated will be deeply re-sequenced in patients and controls to identify potentially functional polymorphisms explaining association as well as additional rare, penetrant mutations in the same gene. The goal is to sequence cases and controls for the exons and nearby evolutionarily conserved sequence of each gene to identify risk alleles when compared to control patients. b) targeted RNA interference screens in epithelial and immune cells to place candidate genes in known pathways c) microarray analysis of peripheral blood and intestinal biopsy samples from Crohn's disease patients to examine gene expression changes associated with identified causal variants d) functional analysis of domains surrounding causal variants; for example, nucleotide-binding oligomerization domain protein 2 (NOD2) and leucine-rich repeat (LRR) domains e) bioinformatics approaches to identify interactions between pathways f) use of model organisms, such as zebra fish and worms, to elucidate microbe–host cell interactions.
References
-
- Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584–9. - PubMed
-
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. - PubMed
-
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. - PubMed
-
- Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999;22:139–44. - PubMed
-
- Wang DG, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–82. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
