

Simultaneous analysis of all SNPs in genome-wide and re-sequencing association studies
- PMID: 18654633
- PMCID: PMC2464715
- DOI: 10.1371/journal.pgen.1000130
Simultaneous analysis of all SNPs in genome-wide and re-sequencing association studies
Abstract
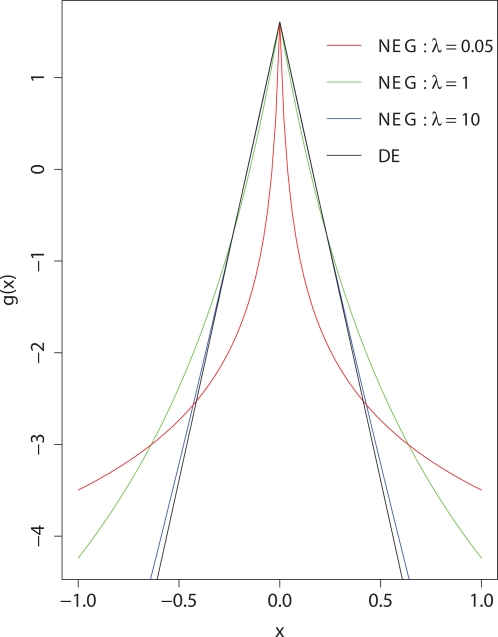
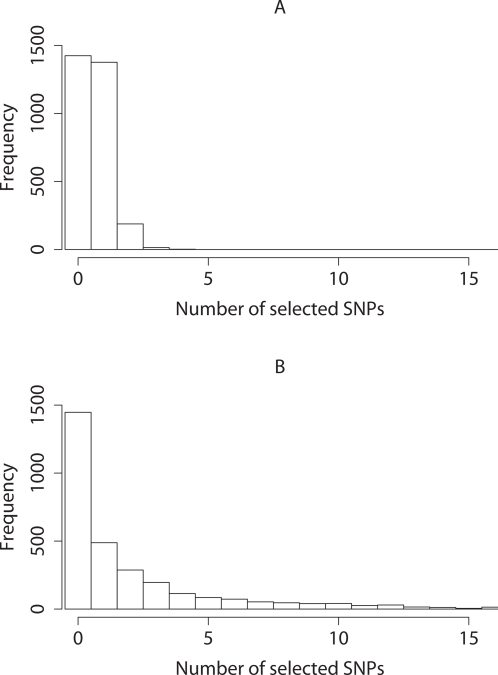
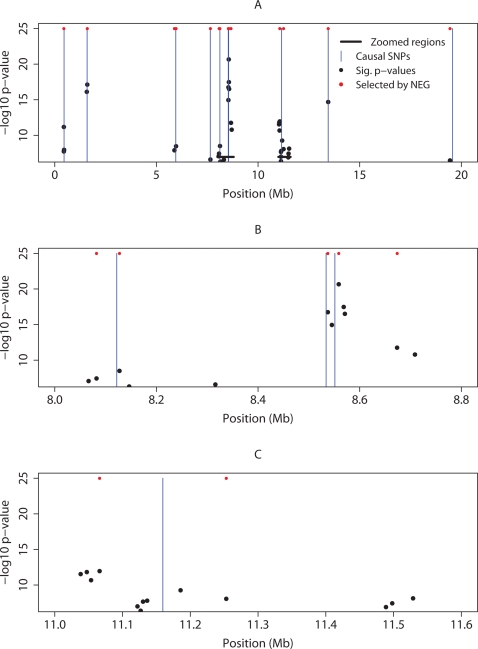
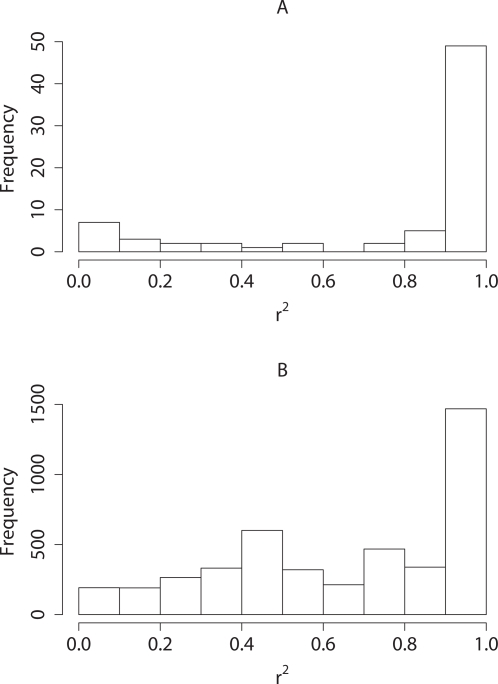
Testing one SNP at a time does not fully realise the potential of genome-wide association studies to identify multiple causal variants, which is a plausible scenario for many complex diseases. We show that simultaneous analysis of the entire set of SNPs from a genome-wide study to identify the subset that best predicts disease outcome is now feasible, thanks to developments in stochastic search methods. We used a Bayesian-inspired penalised maximum likelihood approach in which every SNP can be considered for additive, dominant, and recessive contributions to disease risk. Posterior mode estimates were obtained for regression coefficients that were each assigned a prior with a sharp mode at zero. A non-zero coefficient estimate was interpreted as corresponding to a significant SNP. We investigated two prior distributions and show that the normal-exponential-gamma prior leads to improved SNP selection in comparison with single-SNP tests. We also derived an explicit approximation for type-I error that avoids the need to use permutation procedures. As well as genome-wide analyses, our method is well-suited to fine mapping with very dense SNP sets obtained from re-sequencing and/or imputation. It can accommodate quantitative as well as case-control phenotypes, covariate adjustment, and can be extended to search for interactions. Here, we demonstrate the power and empirical type-I error of our approach using simulated case-control data sets of up to 500 K SNPs, a real genome-wide data set of 300 K SNPs, and a sequence-based dataset, each of which can be analysed in a few hours on a desktop workstation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Genkin A, Lewis DD, Madigan D. Large-scale Bayesian logistic regression for text categorization. Technometrics. 2007;49(3):291–304.
-
- Griffin JE, Brown PJ. Bayesian adaptive Lassos with non–convex penalization. 2007. Technical report, University of Kent.
-
- Breiman L. Heuristics of instability and stabilization in model selection. Annals of Statistics. 1996;24:2350–38.
-
- Mitchel TJ, Beauchamp JJ. Bayesian variable selection in linear regression. J Am Stat Ass. 1988;83:1023–1032.
-
- George EI, McCulloch RI. Variable selection via Gibbs sampling. J Am Stat Ass. 1993;88:881–889.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
