

Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis
- PMID: 18658109
- PMCID: PMC2577727
- DOI: 10.1164/rccm.200712-1804OC
Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis
Abstract
Rationale: The effectiveness and safety of aztreonam lysine for inhalation (AZLI) in patients with cystic fibrosis (CF) on maintenance treatment for Pseudomonas aeruginosa (PA) airway infection was evaluated in this randomized, double-blind, placebo-controlled study.
Objectives: To evaluate the safety and efficacy of inhaled aztreonam lysine in controlling PA infection in patients with CF.
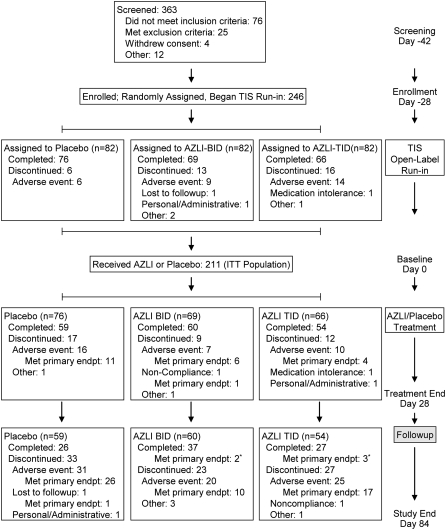
Methods: After randomization and a 28-day course of tobramycin inhalation solution (TIS), patients (n = 211; > or =6 yr; > or =3 TIS courses within previous year; FEV(1) > or = 25% and < or =75% predicted values) were treated with 75 mg AZLI or placebo, twice or three times daily for 28 days, then monitored for 56 days. The primary efficacy endpoint was time to need for additional inhaled or intravenous antipseudomonal antibiotics. Secondary endpoints included changes in respiratory symptoms (CF Questionnaire-Revised [CFQ-R] Respiratory Scale), pulmonary function (FEV(1)), and sputum PA density. Adverse events and minimum inhibitory concentrations of aztreonam for PA were monitored.
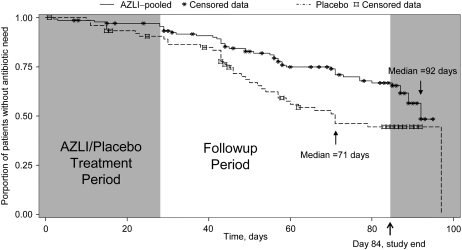
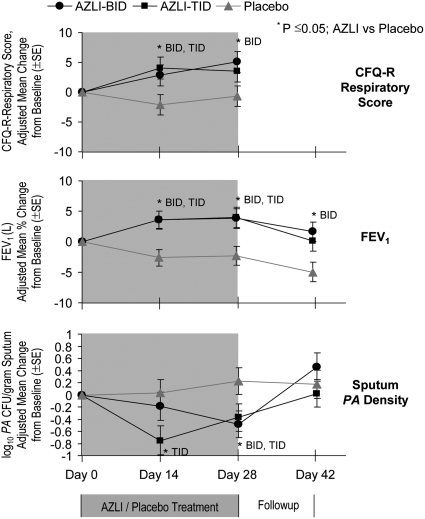
Measurements and main results: AZLI treatment increased median time to need for additional antipseudomonal antibiotics for symptoms of pulmonary exacerbation by 21 days, compared with placebo (AZLI, 92 d; placebo, 71 d; P = 0.007). AZLI improved mean CFQ-R respiratory scores (5.01 points, P = 0.02), FEV(1) (6.3%, P = 0.001), and sputum PA density (-0.66 log(10) cfu/g, P = 0.006) compared with placebo; no AZLI dose-response was observed. Adverse events reported for AZLI and placebo were comparable and consistent with CF lung disease. Susceptibility of PA to aztreonam at baseline and end of therapy were similar.
Conclusions: AZLI was effective in patients with CF using frequent TIS therapy. AZLI delayed time to need for inhaled or intravenous antipseudomonal antibiotics, improved respiratory symptoms and pulmonary function, and was well tolerated. Clinical trial registered with www.clinicaltrials.gov (NCT 00104520).
Trial registration: ClinicalTrials.gov NCT00104520.
Figures
References
-
- Gibson RL, Burns JL, Ramsey BW. State of the art: pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003;168:918–951. - PubMed
-
- Cystic Fibrosis Foundation Patient Registry. 2005 annual data report to the center directors. Bethesda, MD: Cystic Fibrosis Foundation; 2006.
-
- Pamukcu A, Bush A, Buchdahl R. Effects of Pseudomonas aeruginosa colonization on lung function and anthropometric variables in children with cystic fibrosis. Pediatr Pulmonol 1995;19:10–15. - PubMed
-
- Henry RL, Mellis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 1992;12:158–161. - PubMed
-
- Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev-KM, Borowitz D, Bowman CM, Marshall BC, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N Engl J Med 1999;341:23–30. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
