

Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease
- PMID: 18661169
- PMCID: PMC2683920
- DOI: 10.1007/s00246-008-9267-3
Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease
Abstract
Background: Infantile Pompe disease (glycogen storage disease type 2) is a fatal disorder caused by deficiency of acid alpha-glucosidase. This deficiency results in glycogen accumulation in the lysosomes of many tissues including cardiac muscle. The disease is characterized by profound hypotonia, poor growth, organomegaly, and cardiomegaly. Severe hypertrophic cardiomyopathy often is present in early infancy, and most patients die of cardiac or respiratory failure in the first year of life. This report describes the cardiac response of infants with Pompe disease to a phase 2 trial of enzyme replacement therapy (ERT).
Methods: Eight patients with classical infantile Pompe disease were given intravenous recombinant human GAA (rhGAA) for 1 year. Cardiac monitoring included echocardiography, electrocardiograms (ECGs), chest radiographs, and clinical cardiac evaluation at 4, 8, 12, 24, 36, and 52 weeks. At 52 weeks, 6 patients were alive.
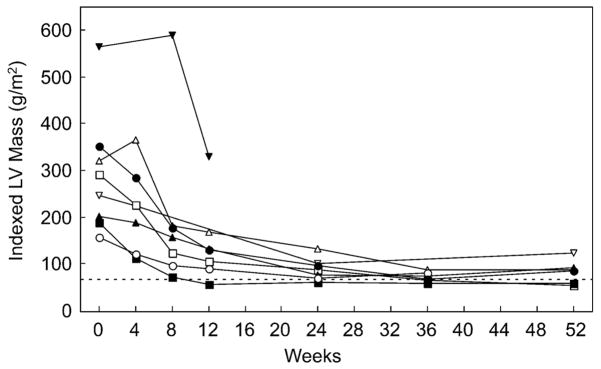
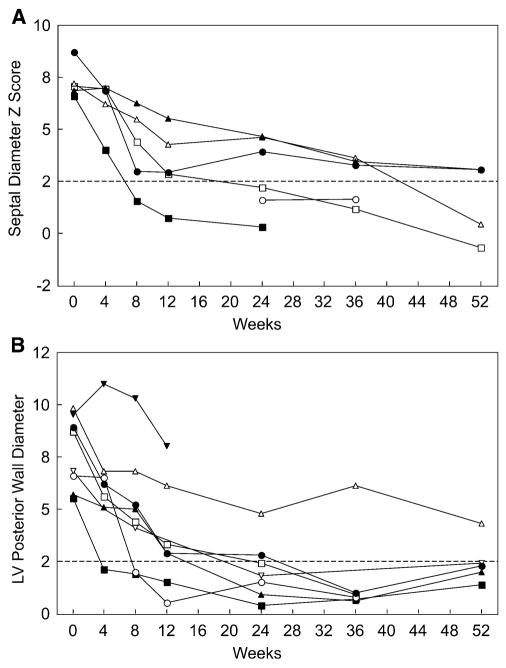
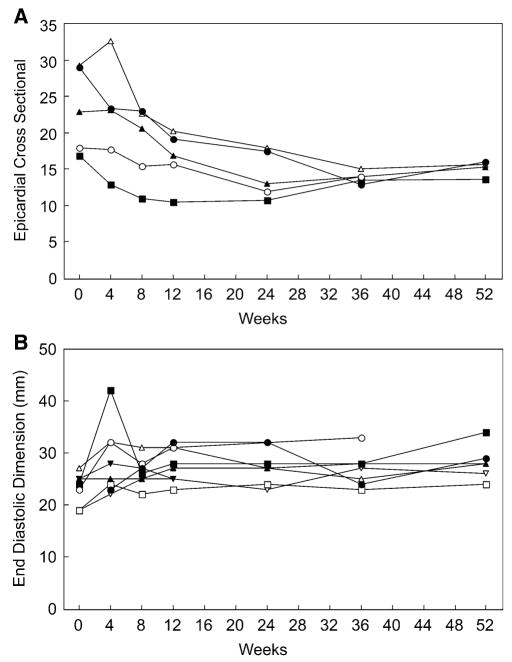
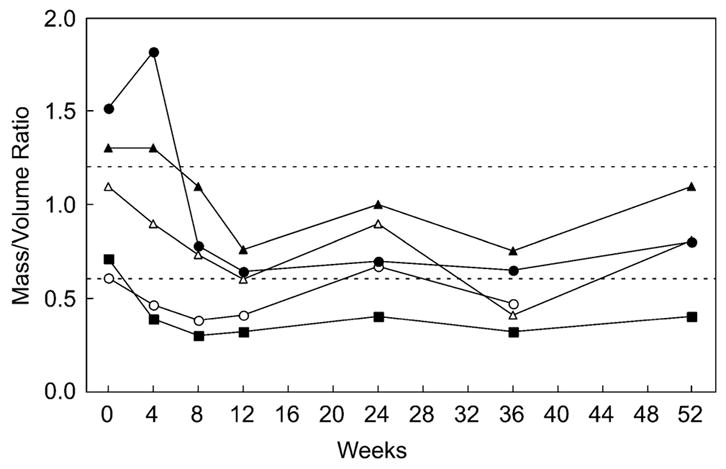
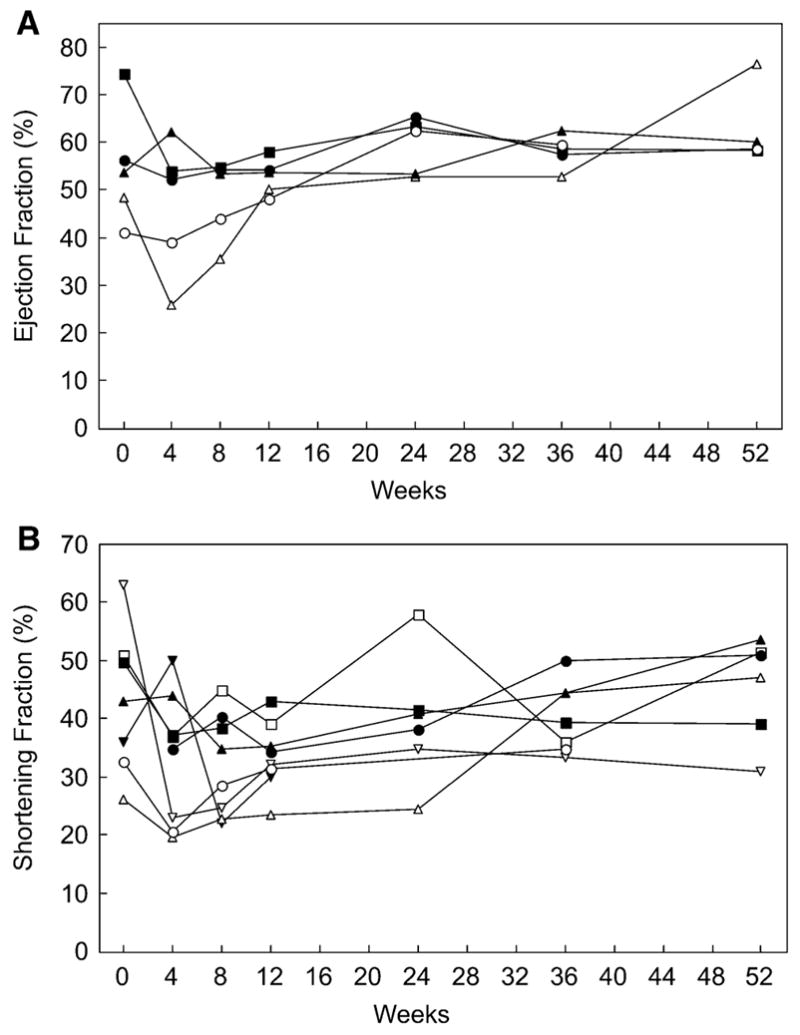
Results: Most of the treated patients had rapid regression of ventricular hypertrophy in response to ERT, with near normalization of posterior wall thickness, ventricular mass, and ventricular size. Systolic ventricular function was preserved despite rapid changes in ventricular mass and size. Concomitantly, ECGs documented lengthening of the PR interval and decreased ventricular voltages, whereas chest radiographs documented a decreased cardiothoracic ratio. Symptoms of pulmonary congestion were diminished, and survival was improved.
Conclusion: The cardiovascular system responds quickly and strikingly to ERT with rhGAA, suggesting rapid reversal of excessive glycogen storage in cardiac muscle cells. Changes in ventricular mass and function are maintained throughout 1 year of follow-up evaluation and associated with decreased morbidity and prolonged survival.
Figures
References
-
- Kishnani P, Howell RR. Pompe disease in infants and children. J Pediatrics. 2004;144:S35–S43. - PubMed
-
- Griffin JL. Infantile acid maltase deficiency: muscle fiber hypertrophy and the ultrastructure of end-stage fibers. Virchows Arch Cell Pathol. 1984;45:37–50. - PubMed
-
- Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Gen. 1998;79:69–72. - PubMed
-
- Van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe disease: 20 original cases compared with 133 cases from literature. Pediatrics. 2003;112:332–340. - PubMed
-
- Kishnani PS, Hwu WL, Mandel H, et al. A retrospective multinational multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
