

Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition
- PMID: 18661558
- PMCID: PMC10103738
- DOI: 10.1002/ana.21449
Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition
Abstract
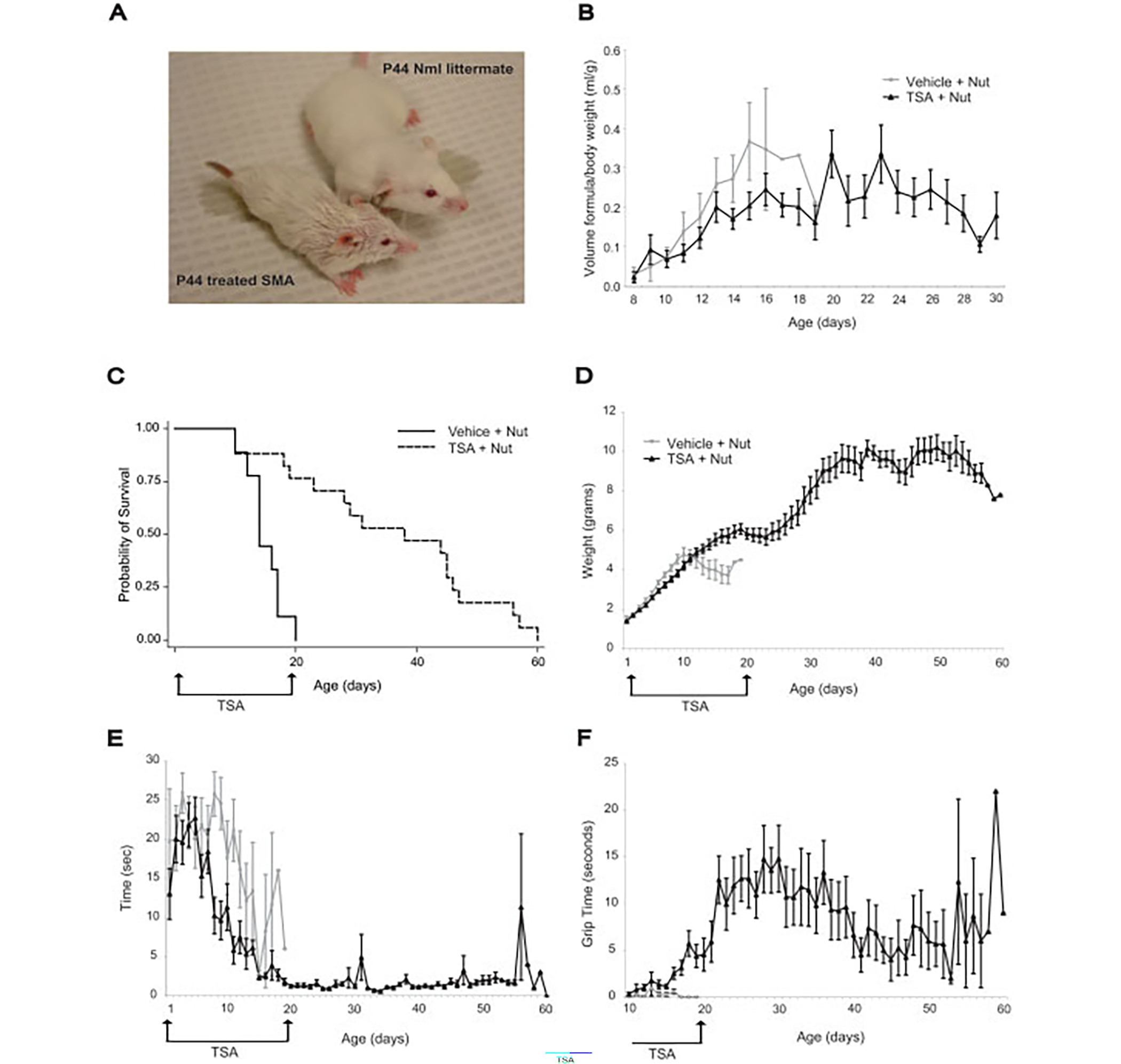
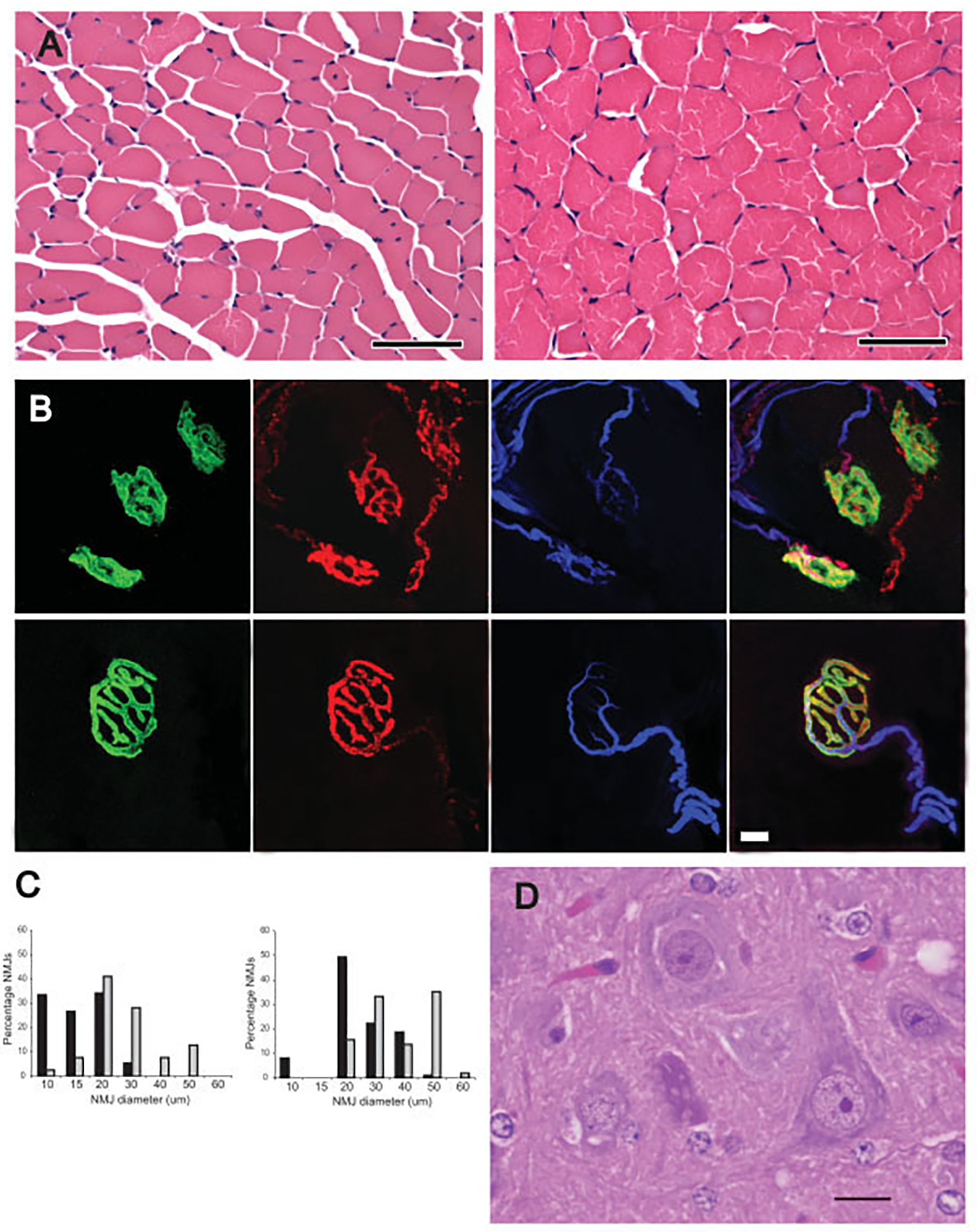
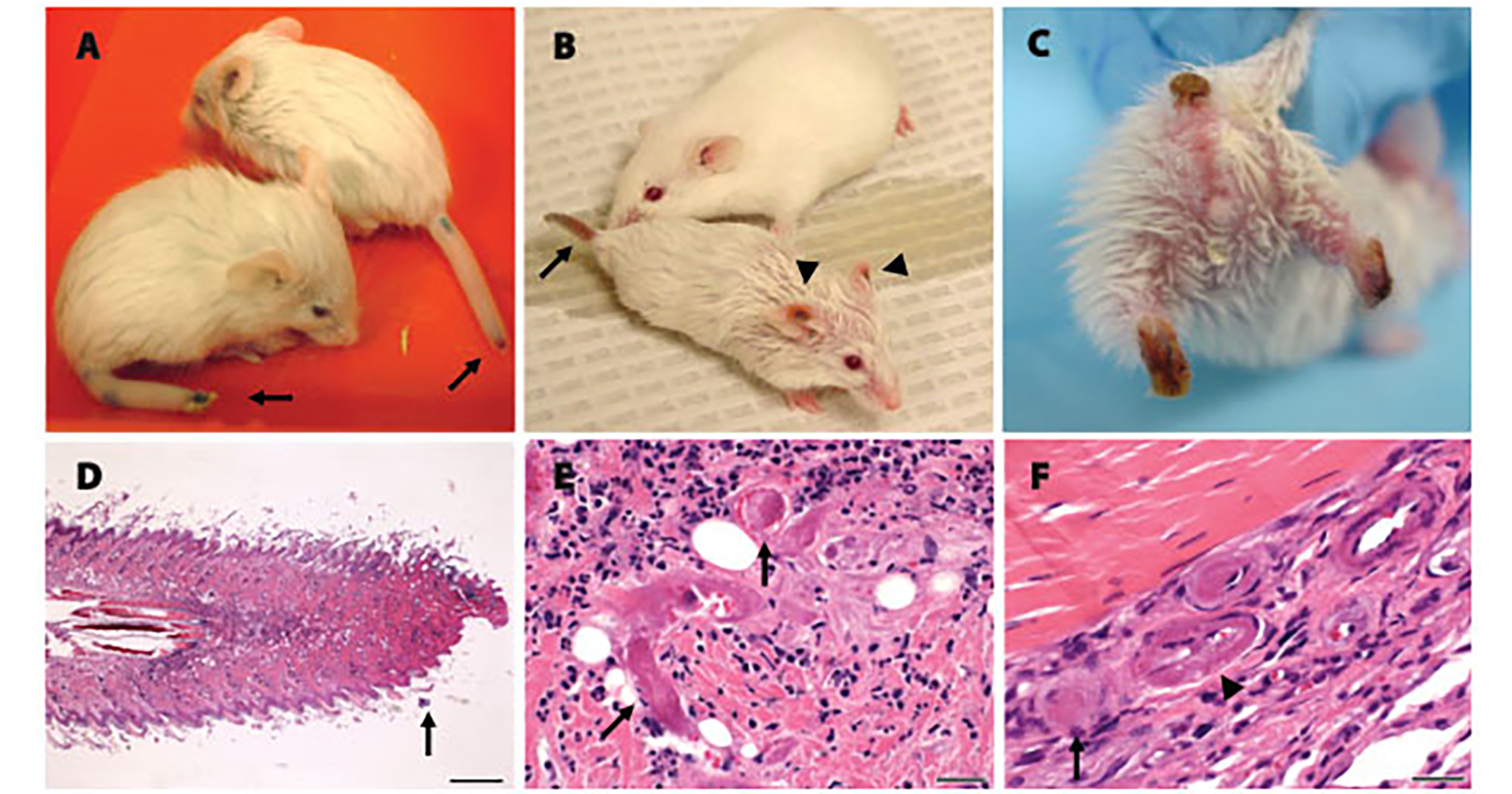
Early treatment with the histone deacetylase inhibitor, trichostatin A, plus nutritional support extended median survival of spinal muscular atrophy mice by 170%. Treated mice continued to gain weight, maintained stable motor function, and retained intact neuromuscular junctions long after trichostatin A was discontinued. In many cases, ultimate decline of mice appeared to result from vascular necrosis, raising the possibility that vascular dysfunction is part of the clinical spectrum of severe spinal muscular atrophy. Early spinal muscular atrophy disease detection and treatment initiation combined with aggressive ancillary care may be integral to the optimization of histone deacetylase inhibitor treatment in human patients.
Figures
References
-
- Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165. - PubMed
-
- Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999;8:1177–1183. - PubMed
-
- Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997;16:265–269. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
