

ACTH inhibits bTREK-1 K+ channels through multiple cAMP-dependent signaling pathways
- PMID: 18663135
- PMCID: PMC2483331
- DOI: 10.1085/jgp.200810003
ACTH inhibits bTREK-1 K+ channels through multiple cAMP-dependent signaling pathways
Abstract
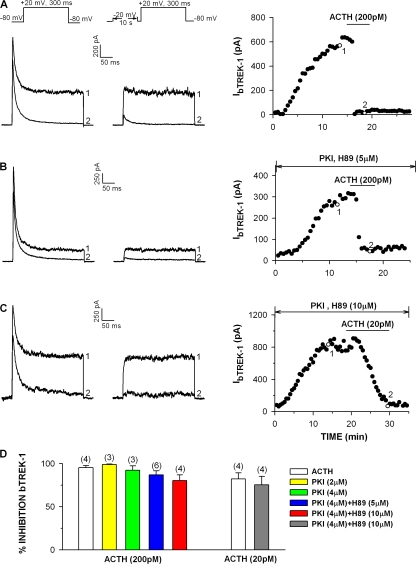
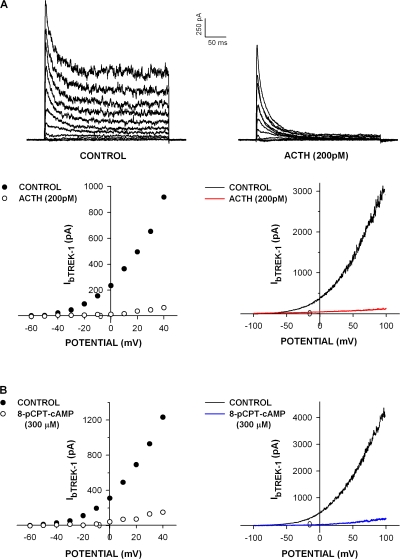
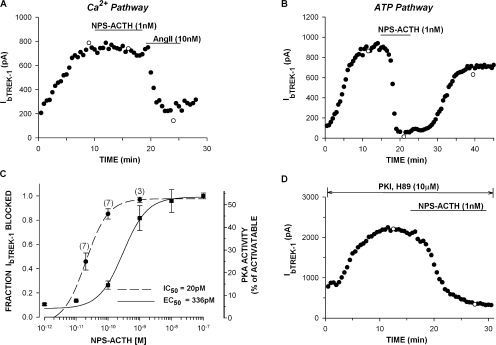
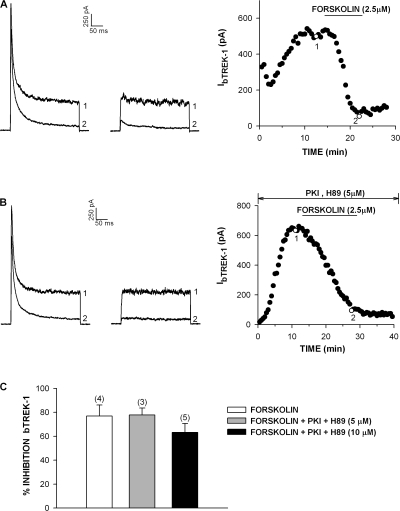
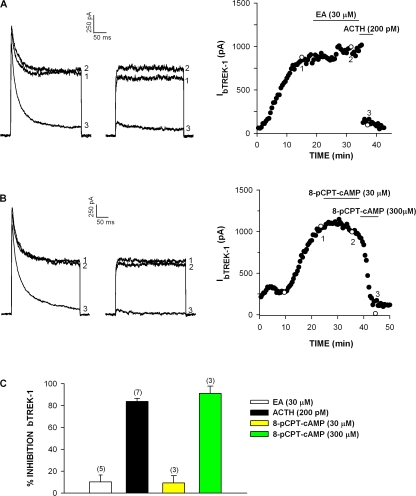
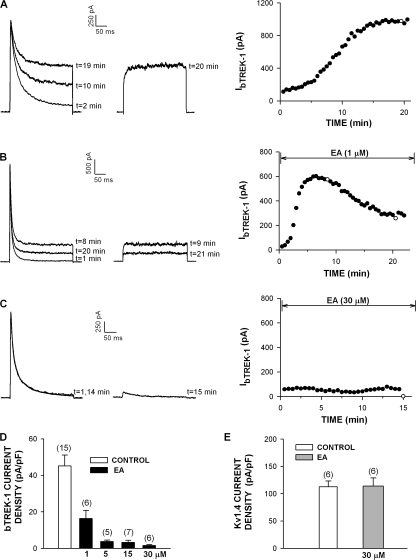
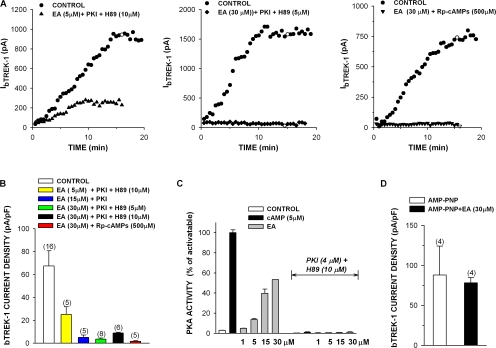
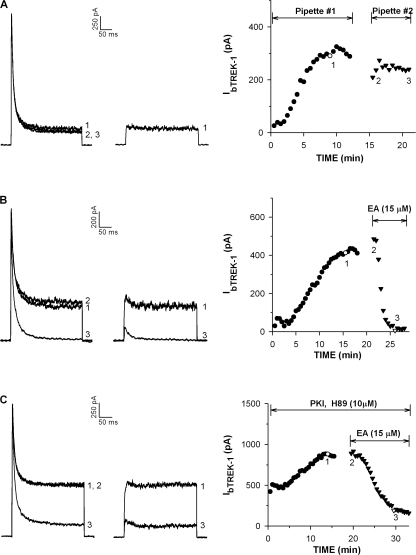
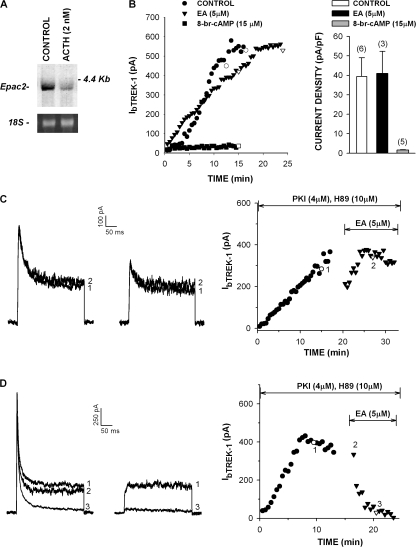
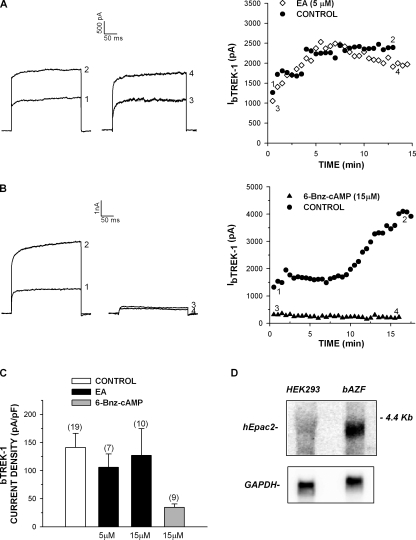
Bovine adrenal zona fasciculata (AZF) cells express bTREK-1 K(+) channels that set the resting membrane potential and function pivotally in the physiology of cortisol secretion. Inhibition of these K(+) channels by adrenocorticotropic hormone (ACTH) or cAMP is coupled to depolarization and Ca(2+) entry. The mechanism of ACTH and cAMP-mediated inhibition of bTREK-1 was explored in whole cell patch clamp recordings from AZF cells. Inhibition of bTREK-1 by ACTH and forskolin was not affected by the addition of both H-89 and PKI (6-22) amide to the pipette solution at concentrations that completely blocked activation of cAMP-dependent protein kinase (PKA) in these cells. The ACTH derivative, O-nitrophenyl, sulfenyl-adrenocorticotropin (NPS-ACTH), at concentrations that produced little or no activation of PKA, inhibited bTREK-1 by a Ca(2+)-independent mechanism. Northern blot analysis showed that bovine AZF cells robustly express mRNA for Epac2, a guanine nucleotide exchange protein activated by cAMP. The selective Epac activator, 8-pCPT-2'-O-Me-cAMP, applied intracellularly through the patch pipette, inhibited bTREK-1 (IC(50) = 0.63 microM) at concentrations that did not activate PKA. Inhibition by this agent was unaffected by PKA inhibitors, including RpcAMPS, but was eliminated in the absence of hydrolyzable ATP. Culturing AZF cells in the presence of ACTH markedly reduced the expression of Epac2 mRNA. 8-pCPT-2'-O-Me-cAMP failed to inhibit bTREK-1 current in AZF cells that had been treated with ACTH for 3-4 d while inhibition by 8-br-cAMP was not affected. 8-pCPT-2'-O-Me-cAMP failed to inhibit bTREK-1 expressed in HEK293 cells, which express little or no Epac2. These findings demonstrate that, in addition to the well-described PKA-dependent TREK-1 inhibition, ACTH, NPS-ACTH, forskolin, and 8-pCPT-2'-O-Me-cAMP also inhibit these K(+) channels by a PKA-independent signaling pathway. The convergent inhibition of bTREK-1 through parallel PKA- and Epac-dependent mechanisms may provide for failsafe membrane depolarization by ACTH.
Figures
Similar articles
-
cAMP analogs and their metabolites enhance TREK-1 mRNA and K+ current expression in adrenocortical cells.Mol Pharmacol. 2010 Mar;77(3):469-82. doi: 10.1124/mol.109.061861. Epub 2009 Dec 22. Mol Pharmacol. 2010. PMID: 20028740 Free PMC article.
-
N6-substituted cAMP analogs inhibit bTREK-1 K+ channels and stimulate cortisol secretion by a protein kinase A-independent mechanism.Mol Pharmacol. 2009 Dec;76(6):1290-301. doi: 10.1124/mol.109.057075. Epub 2009 Sep 4. Mol Pharmacol. 2009. PMID: 19734321 Free PMC article.
-
Evidence for cAMP-independent bTREK-1 inhibition by ACTH and NPS-ACTH in adrenocortical cells.Mol Cell Endocrinol. 2012 Jan 2;348(1):305-12. doi: 10.1016/j.mce.2011.09.023. Epub 2011 Sep 17. Mol Cell Endocrinol. 2012. PMID: 21952081
-
Biochemical and Ionic signaling mechanisms for ACTH-stimulated cortisol production.Vitam Horm. 2005;70:265-79. doi: 10.1016/S0083-6729(05)70008-X. Vitam Horm. 2005. PMID: 15727807 Review.
-
Cell physiology of cAMP sensor Epac.J Physiol. 2006 Nov 15;577(Pt 1):5-15. doi: 10.1113/jphysiol.2006.119644. Epub 2006 Sep 14. J Physiol. 2006. PMID: 16973695 Free PMC article. Review.
Cited by
-
Role of EPAC in cAMP-Mediated Actions in Adrenocortical Cells.Front Endocrinol (Lausanne). 2016 Jun 13;7:63. doi: 10.3389/fendo.2016.00063. eCollection 2016. Front Endocrinol (Lausanne). 2016. PMID: 27379015 Free PMC article. Review.
-
NCI-H295R, a human adrenal cortex-derived cell line, expresses purinergic receptors linked to Ca²⁺-mobilization/influx and cortisol secretion.PLoS One. 2013 Aug 8;8(8):e71022. doi: 10.1371/journal.pone.0071022. eCollection 2013. PLoS One. 2013. PMID: 23951072 Free PMC article.
-
Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development.Physiol Rev. 2018 Apr 1;98(2):919-1053. doi: 10.1152/physrev.00025.2017. Physiol Rev. 2018. PMID: 29537337 Free PMC article. Review.
-
cAMP analogs and their metabolites enhance TREK-1 mRNA and K+ current expression in adrenocortical cells.Mol Pharmacol. 2010 Mar;77(3):469-82. doi: 10.1124/mol.109.061861. Epub 2009 Dec 22. Mol Pharmacol. 2010. PMID: 20028740 Free PMC article.
-
Protein Kinase Inhibitor Peptide as a Tool to Specifically Inhibit Protein Kinase A.Front Physiol. 2020 Nov 25;11:574030. doi: 10.3389/fphys.2020.574030. eCollection 2020. Front Physiol. 2020. PMID: 33324237 Free PMC article. Review.
References
-
- Awad, J.A., R.A. Johnson, K.H. Jakobs, and G. Schultz. 1983. Interactions of forskolin and adenylate cyclase. Effects on substrate kinetics and protection against inactivation by heat and N-ethylmaleimide. J. Biol. Chem. 258:2960–2965. - PubMed
-
- Bockenhauer, D., N. Zilberberg, and S.A. Goldstein. 2001. KCNK2: reversible conversion of a hippocampal potassium leak into a voltage-dependent channel. Nat. Neurosci. 4:486–491. - PubMed
-
- Bos, J.L. 2003. Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 4:733–738. - PubMed
-
- Brooks, S.P., and K.B. Storey. 1992. Bound and determined: a computer program for making buffers of defined ion concentrations. Anal. Biochem. 201:119–126. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
