

Conserved secondary structures in Aspergillus
- PMID: 18665251
- PMCID: PMC2467506
- DOI: 10.1371/journal.pone.0002812
Conserved secondary structures in Aspergillus
Abstract
Background: Recent evidence suggests that the number and variety of functional RNAs (ncRNAs as well as cis-acting RNA elements within mRNAs) is much higher than previously thought; thus, the ability to computationally predict and analyze RNAs has taken on new importance. We have computationally studied the secondary structures in an alignment of six Aspergillus genomes. Little is known about the RNAs present in this set of fungi, and this diverse set of genomes has an optimal level of sequence conservation for observing the correlated evolution of base-pairs seen in RNAs.
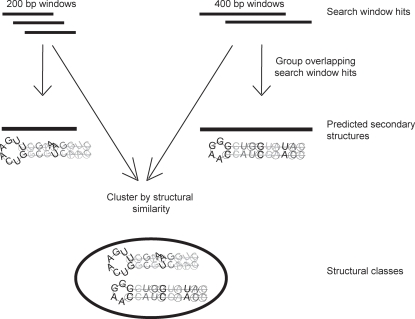
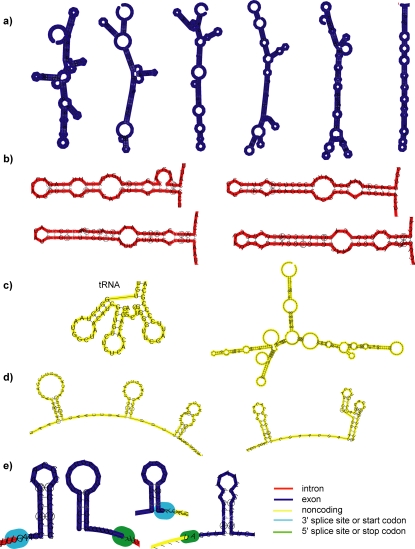
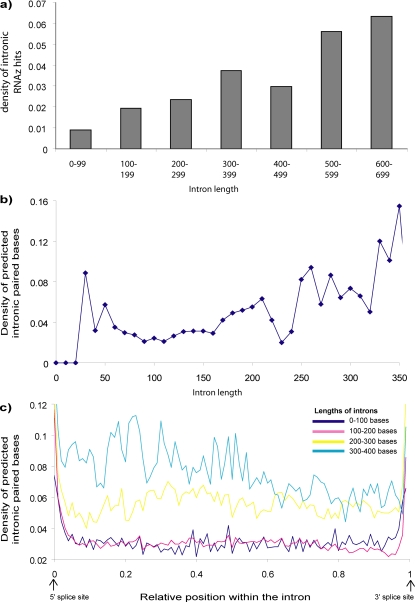
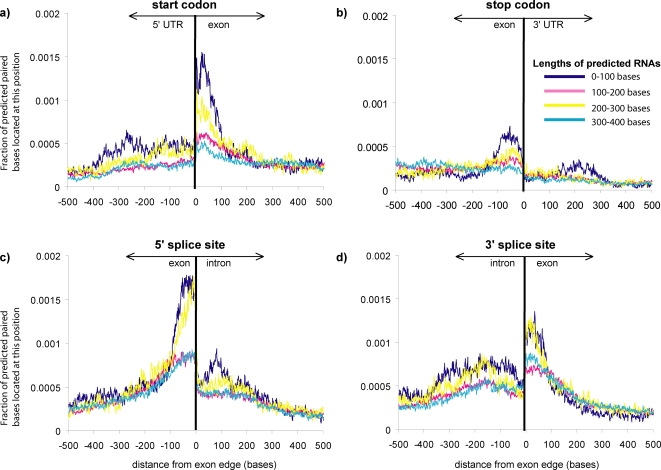
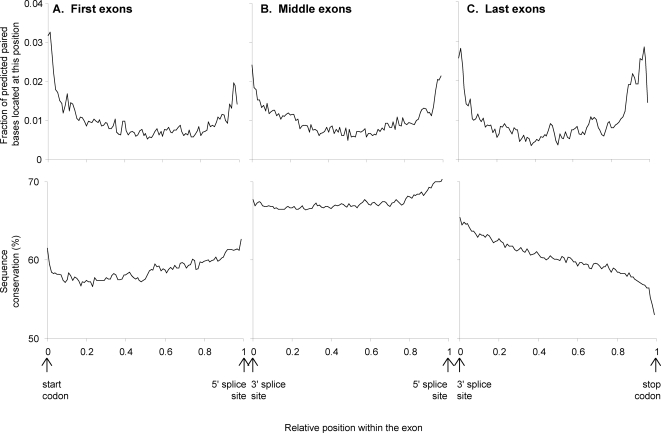
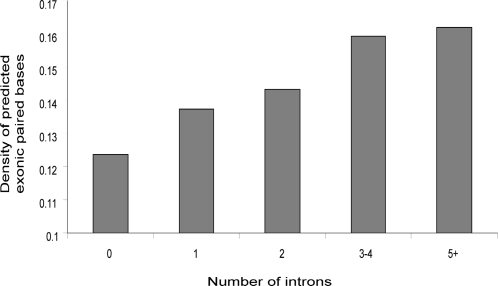
Methodology/principal findings: We report the results of a whole-genome search for evolutionarily conserved secondary structures, as well as the results of clustering these predicted secondary structures by structural similarity. We find a total of 7450 predicted secondary structures, including a new predicted approximately 60 bp long hairpin motif found primarily inside introns. We find no evidence for microRNAs. Different types of genomic regions are over-represented in different classes of predicted secondary structures. Exons contain the longest motifs (primarily long, branched hairpins), 5' UTRs primarily contain groupings of short hairpins located near the start codon, and 3' UTRs contain very little secondary structure compared to other regions. There is a large concentration of short hairpins just inside the boundaries of exons. The density of predicted intronic RNAs increases with the length of introns, and the density of predicted secondary structures within mRNA coding regions increases with the number of introns in a gene.
Conclusions/significance: There are many conserved, high-confidence RNAs of unknown function in these Aspergillus genomes, as well as interesting spatial distributions of predicted secondary structures. This study increases our knowledge of secondary structure in these aspergillus organisms.
Conflict of interest statement
Figures
Similar articles
-
Discovery of 17 conserved structural RNAs in fungi.Nucleic Acids Res. 2021 Jun 21;49(11):6128-6143. doi: 10.1093/nar/gkab355. Nucleic Acids Res. 2021. PMID: 34086938 Free PMC article.
-
Detection of RNA structures in porcine EST data and related mammals.BMC Genomics. 2007 Sep 10;8:316. doi: 10.1186/1471-2164-8-316. BMC Genomics. 2007. PMID: 17845718 Free PMC article.
-
Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome.Nat Biotechnol. 2005 Nov;23(11):1383-90. doi: 10.1038/nbt1144. Nat Biotechnol. 2005. PMID: 16273071
-
The structure and functions of coronavirus genomic 3' and 5' ends.Virus Res. 2015 Aug 3;206:120-33. doi: 10.1016/j.virusres.2015.02.025. Epub 2015 Feb 28. Virus Res. 2015. PMID: 25736566 Free PMC article. Review.
-
Splicing noncoding RNAs from the inside out.Wiley Interdiscip Rev RNA. 2015 Nov-Dec;6(6):651-60. doi: 10.1002/wrna.1307. Epub 2015 Oct 1. Wiley Interdiscip Rev RNA. 2015. PMID: 26424453 Free PMC article. Review.
Cited by
-
Known and novel post-transcriptional regulatory sequences are conserved across plant families.RNA. 2012 Mar;18(3):368-84. doi: 10.1261/rna.031179.111. Epub 2012 Jan 11. RNA. 2012. PMID: 22237150 Free PMC article.
-
Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei.PLoS Negl Trop Dis. 2013 Aug 22;7(8):e2398. doi: 10.1371/journal.pntd.0002398. eCollection 2013. PLoS Negl Trop Dis. 2013. PMID: 23991243 Free PMC article.
References
-
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. - PubMed
-
- Johnson JM, Edwards S, Shoemaker D, Schadt EE. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005;21:93–102. - PubMed
-
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. - PubMed
-
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
