

Gitelman syndrome
- PMID: 18667063
- PMCID: PMC2518128
- DOI: 10.1186/1750-1172-3-22
Gitelman syndrome
Abstract
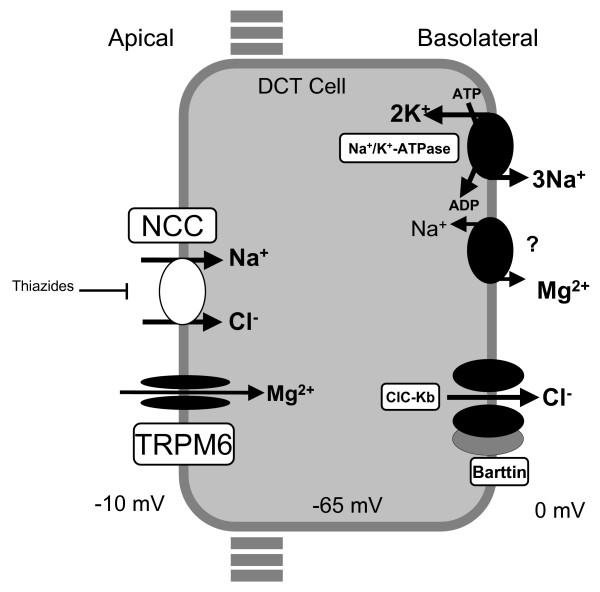
Gitelman syndrome (GS), also referred to as familial hypokalemia-hypomagnesemia, is characterized by hypokalemic metabolic alkalosis in combination with significant hypomagnesemia and low urinary calcium excretion. The prevalence is estimated at approximately 1:40,000 and accordingly, the prevalence of heterozygotes is approximately 1% in Caucasian populations, making it one of the most frequent inherited renal tubular disorders. In the majority of cases, symptoms do not appear before the age of six years and the disease is usually diagnosed during adolescence or adulthood. Transient periods of muscle weakness and tetany, sometimes accompanied by abdominal pain, vomiting and fever are often seen in GS patients. Paresthesias, especially in the face, frequently occur. Remarkably, some patients are completely asymptomatic except for the appearance at adult age of chondrocalcinosis that causes swelling, local heat, and tenderness over the affected joints. Blood pressure is lower than that in the general population. Sudden cardiac arrest has been reported occasionally. In general, growth is normal but can be delayed in those GS patients with severe hypokalemia and hypomagnesemia.GS is transmitted as an autosomal recessive trait. Mutations in the solute carrier family12, member 3 gene, SLC12A3, which encodes the thiazide-sensitive NaCl cotransporter (NCC), are found in the majority of GS patients. At present, more than 140 different NCC mutations throughout the whole protein have been identified. In a small minority of GS patients, mutations in the CLCNKB gene, encoding the chloride channel ClC-Kb have been identified.Diagnosis is based on the clinical symptoms and biochemical abnormalities (hypokalemia, metabolic alkalosis, hypomagnesemia and hypocalciuria). Bartter syndrome (especially type III) is the most important genetic disorder to consider in the differential diagnosis of GS. Genetic counseling is important. Antenatal diagnosis for GS is technically feasible but not advised because of the good prognosis in the majority of patients.Most asymptomatic patients with GS remain untreated and undergo ambulatory monitoring, once a year, generally by nephrologists. Lifelong supplementation of magnesium (magnesium-oxide and magnesium-sulfate) is recommended. Cardiac work-up should be offered to screen for risk factors of cardiac arrhythmias. All GS patients are encouraged to maintain a high-sodium and high potassium diet. In general, the long-term prognosis of GS is excellent.
Figures
References
-
- Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235. - PubMed
-
- Riveira-Munoz E, Chang Q, Godefroid N, Hoenderop JG, Bindels RJ, Dahan K, Devuyst O. Belgian network for study of gitelman syndrome. Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol. 2007;1:1271–1283. doi: 10.1681/ASN.2006101095. - DOI - PubMed
-
- Scognamiglio R, Negut C, Calò LA. Aborted sudden cardiac death in two patients with Bartter's/Gitelman's syndromes. Clin Nephrol. 2007;67:193–197. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
