

A low-molecular-weight antagonist for the human thyrotropin receptor with therapeutic potential for hyperthyroidism
- PMID: 18669595
- PMCID: PMC2613050
- DOI: 10.1210/en.2008-0836
A low-molecular-weight antagonist for the human thyrotropin receptor with therapeutic potential for hyperthyroidism
Abstract
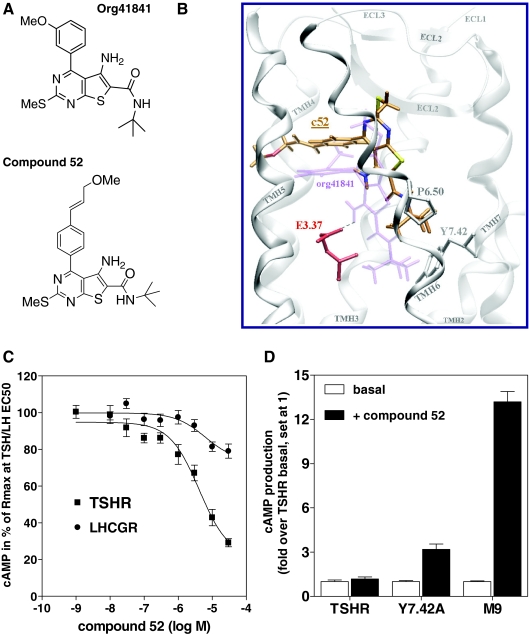
Low-molecular-weight (LMW) antagonists for TSH receptor (TSHR) may have therapeutic potential as orally active drugs to block stimulating antibodies (TsAbs) in Graves' hyperthyroidism. We describe an approach to identify LMW ligands for TSHR based on Org41841, a LMW partial agonist for the LH/choriogonadotropin receptor and TSHR. We used molecular modeling and functional experiments to guide the chemical modification of Org41841. We identified an antagonist (NIDDK/CEB-52) that selectively inhibits activation of TSHR by both TSH and TsAbs. Whereas initially characterized in cultured cells overexpressing TSHRs, the antagonist was also active under more physiologically relevant conditions in primary cultures of human thyrocytes expressing endogenous TSHRs in which it inhibited TSH- and TsAb-induced up-regulation of mRNA transcripts for thyroperoxidase. Our results establish this LMW compound as a lead for the development of higher potency antagonists and serve as proof of principle that LMW ligands that target TSHR could serve as drugs in patients with Graves' disease.
Figures

Comment in
-
Antithyroid drugs are 65 years old: time for retirement?Endocrinology. 2008 Dec;149(12):5943-4. doi: 10.1210/en.2008-1349. Endocrinology. 2008. PMID: 19022900 No abstract available.
References
-
- Ascoli M, Fanelli F, Segaloff DL 2002 The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev 23:141–174 - PubMed
-
- Szkudlinski MW, Fremont V, Ronin C, Weintraub BD 2002 Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev 82:473–502 - PubMed
-
- Vassart G, Pardo L, Costagliola S 2004 A molecular dissection of the glycoprotein hormone receptors. Trends Biochem Sci 29:119–126 - PubMed
-
- Guo T 2005 Small molecule agonists and antagonists for the LH and FSH receptors. Expert Opin Ther Patents 15:1555–1564
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
