

Hamartomatous polyposis syndromes
- PMID: 18672141
- PMCID: PMC2659506
- DOI: 10.1016/j.suc.2008.05.002
Hamartomatous polyposis syndromes
Abstract
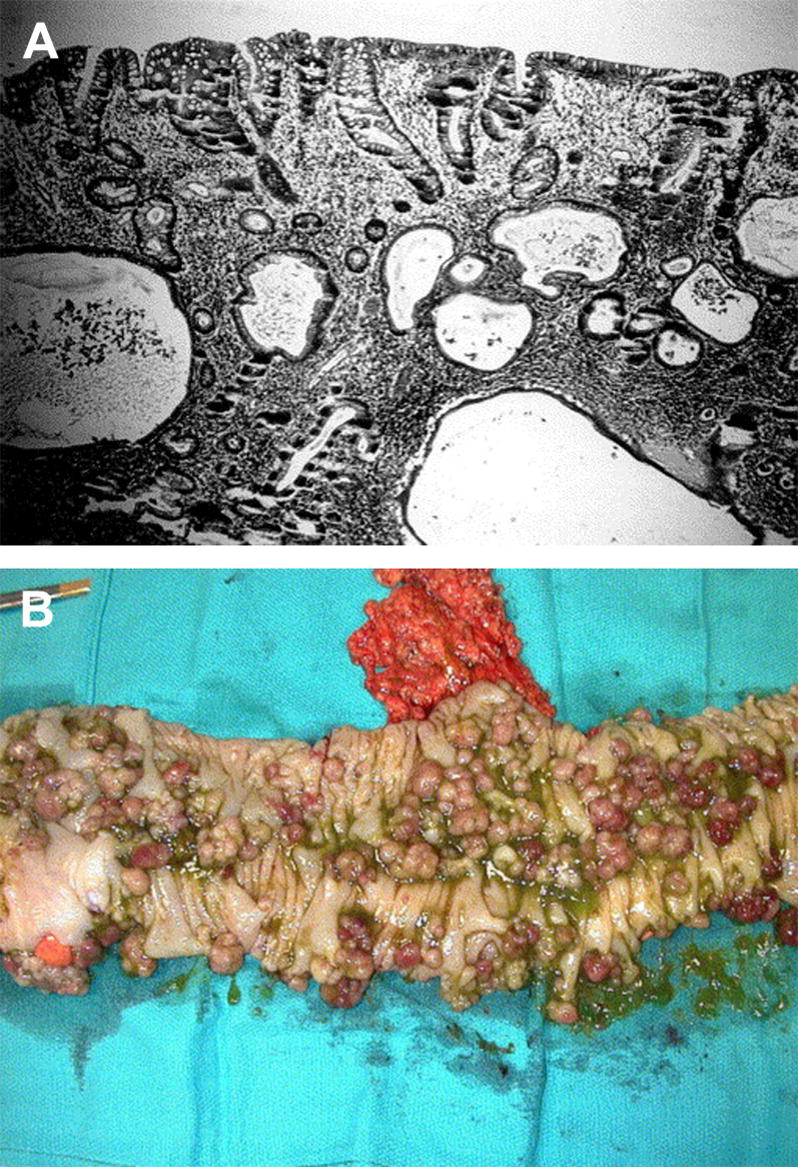
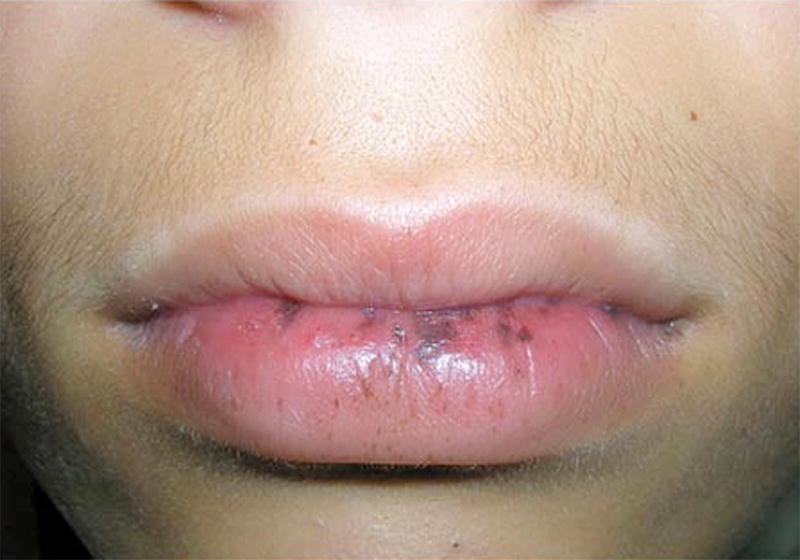
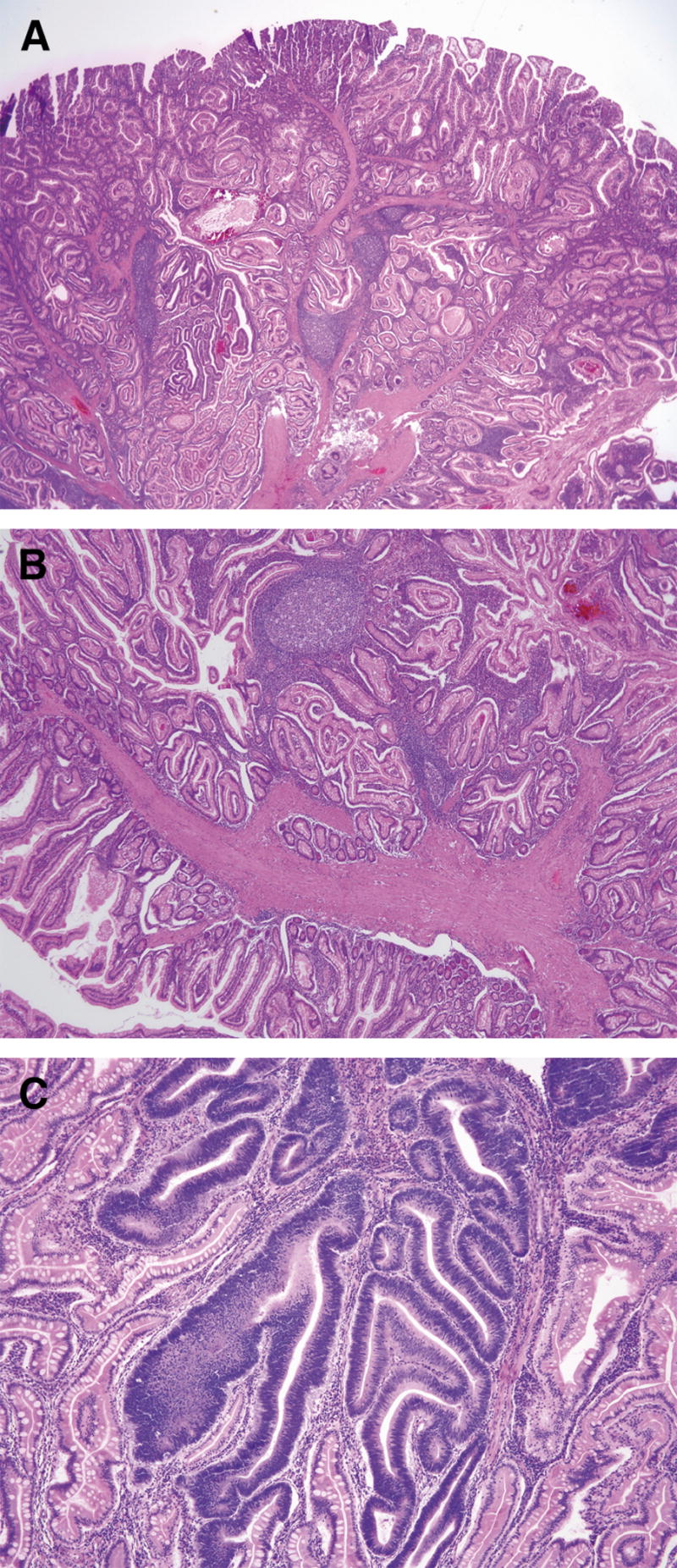
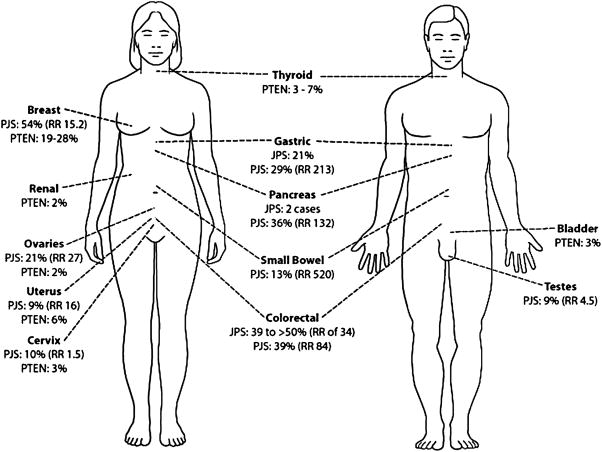
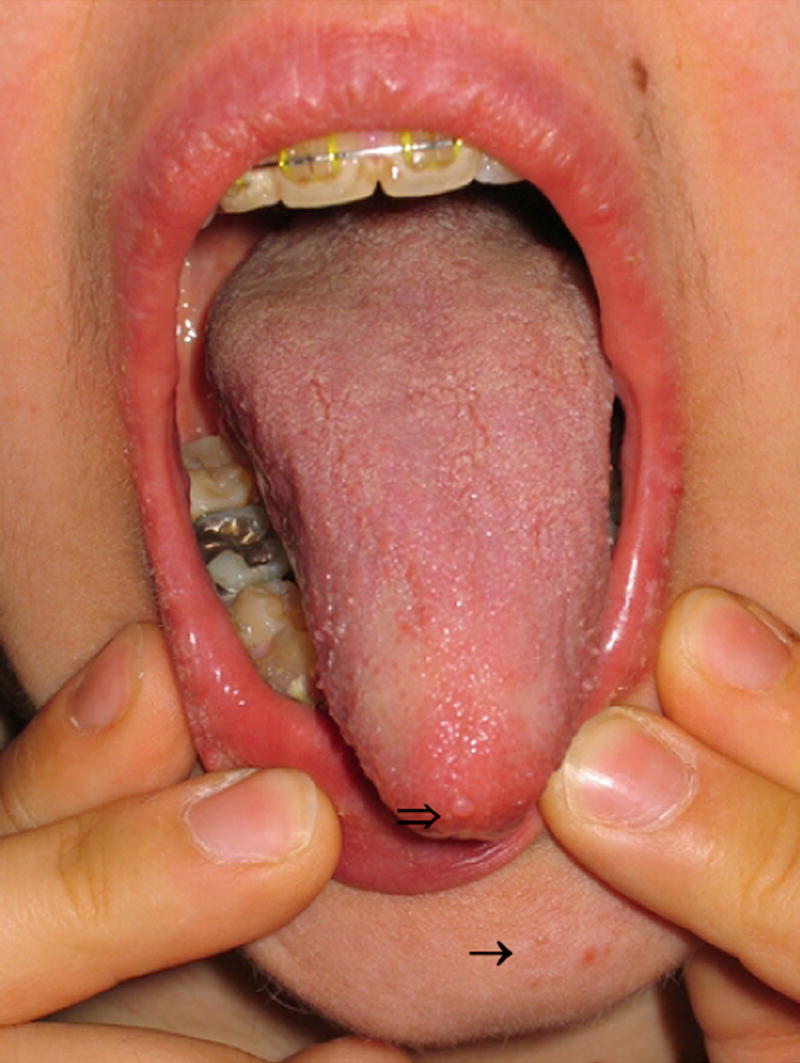
Since the histologic description of the hamartomatous polyp in 1957 by Horrilleno and colleagues, descriptions have appeared of several different syndromes with the propensity to develop these polyps in the upper and lower gastrointestinal tracts. These syndromes include juvenile polyposis, Peutz-Jeghers syndrome, hereditary mixed polyposis syndrome, and the phosphatase and tensin homolog gene (PTEN) hamartoma tumor syndromes (Cowden and Bannayan-Riley-Ruvalcaba syndromes), which are autosomal-dominantly inherited, and Cronkhite-Canada syndrome, which is acquired. This article reviews the clinical aspects, the molecular pathogenesis, the affected organ systems, the risks of cancer, and the management of these hamartomatous polyposis syndromes. Although the incidence of these syndromes is low, it is important for clinicians to recognize these disorders to prevent morbidity and mortality in these patients, and to perform presymptomatic testing in patients at risk.
Figures

References
-
- Albrecht S, Haber RM, Goodman JC, et al. Cowden syndrome and Lhermitte-Duclos disease. Cancer. 1992;70:869. - PubMed
-
- Arch EM, Goodman BK, Van Wesep RA, et al. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with Cowden disease. Am J Med Genet. 1997;71:489. - PubMed
-
- Bannayan GA. Lipomatosis, angiomatosis, and macrencephalia. A previously undescribed congenital syndrome. Arch Pathol. 1971;92:1. - PubMed
-
- Bartholomew LG, More CE, Dahlin DC, et al. Intestinal polyposis associated with mucocutaneous pigmentation. Surgery, Gynecology, and Obstetrics. 1962;115:1. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
