

Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation
- PMID: 18676402
- PMCID: PMC2574026
- DOI: 10.1096/fj.08-112375
Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation
Abstract
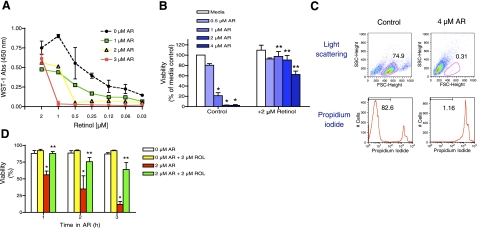
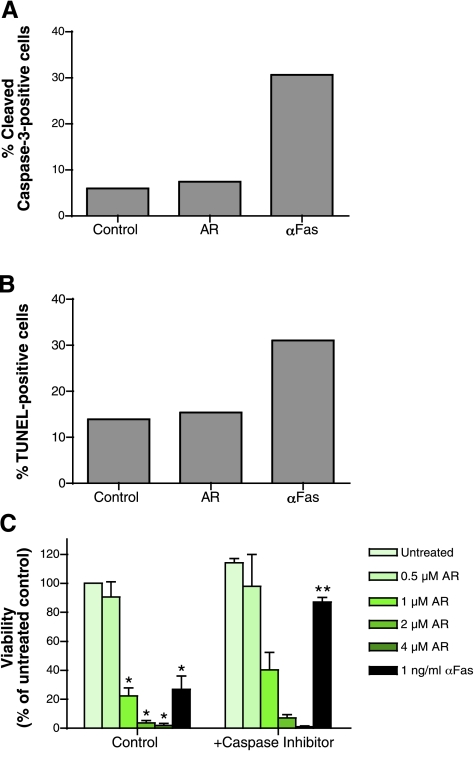
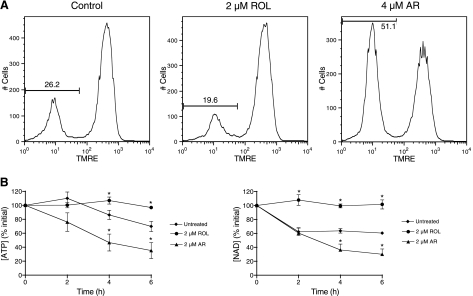
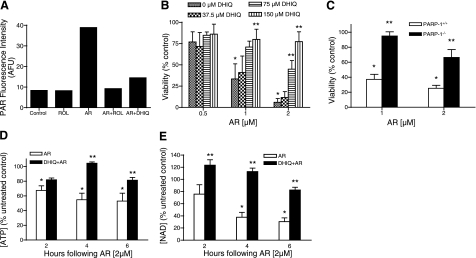
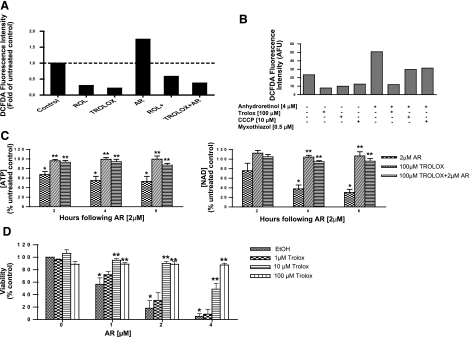
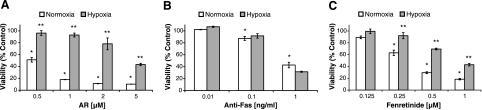
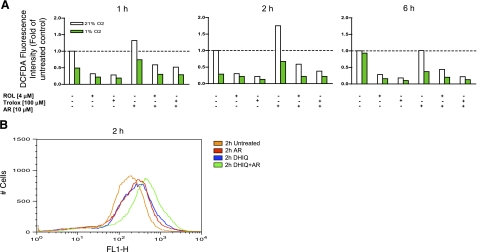
A significant unresolved question is how vitamin A deprivation causes, and why retinoic acid fails to reverse, immunodeficiency. When depleted of vitamin A, T cells undergo programmed cell death (PCD), which is enhanced by the natural competitor of retinol, anhydroretinol. PCD does not happen by apoptosis, despite the occurrence of shared early events, including mitochondrial membrane depolarization, permeability transition pore opening, and cytochrome c release. It also lacks caspase-3 activation, chromatin condensation, and endonuclease-mediated DNA degradation, hallmarks of apoptosis. PCD following vitamin A deprivation exhibits increased production of reactive oxygen species (ROS), drastic reductions in ATP and NAD(+) levels, and activation of poly-(ADP-ribose) polymerase (PARP) -1. These latter steps are causative because neutralizing ROS, imposing hypoxic conditions, or inhibiting PARP-1 by genetic or pharmacologic approaches prevents energy depletion and PCD. The data highlight a novel regulatory role of vitamin A in mitochondrial energy homeostasis.
Figures
References
-
- De Luca L M. Retinoids and their receptors in differentiation, embryogenesis, and neoplasia. FASEB J. 1991;5:2924–2933. - PubMed
-
- Buck J, Derguini F, Levi E, Nakanishi K, Hammerling U. Intracellular signaling by 14-hydroxy-4,14-retro-retinol. Science. 1991;254:1654–1656. - PubMed
-
- Derguini F, Nakanishi K, Hammerling U, Chua R, Eppinger T, Levi E, Buck J. 13,14-Dihydroxy-retinol, a new bioactive retinol metabolite. J Biol Chem. 1995;270:18875–18880. - PubMed
-
- Derguini F, Nakanishi K, Hammerling U, Buck J. Intracellular signaling activity of synthetic (14R)-, (14S)-, and (14RS)-14-hydroxy-4,14-retro-retinol. Biochemistry. 1994;33:623–628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
