

Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair
- PMID: 18676809
- PMCID: PMC2492745
- DOI: 10.1101/gad.472808
Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair
Abstract
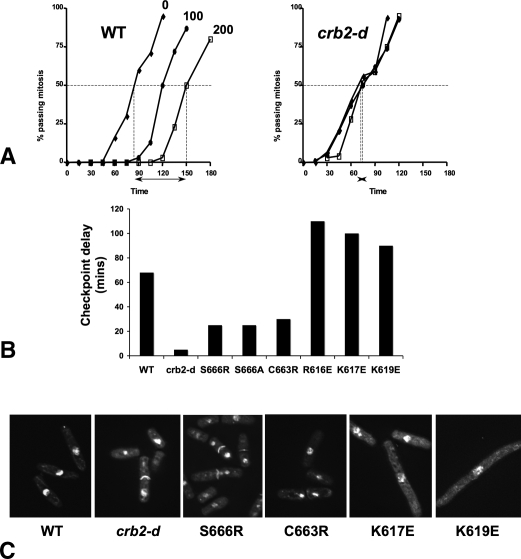
Schizosaccharomyces pombe Crb2 is a checkpoint mediator required for the cellular response to DNA damage. Like human 53BP1 and Saccharomyces cerevisiae Rad9 it contains Tudor(2) and BRCT(2) domains. Crb2-Tudor(2) domain interacts with methylated H4K20 and is required for recruitment to DNA dsDNA breaks. The BRCT(2) domain is required for dimerization, but its precise role in DNA damage repair and checkpoint signaling is unclear. The crystal structure of the Crb2-BRCT(2) domain, alone and in complex with a phosphorylated H2A.1 peptide, reveals the structural basis for dimerization and direct interaction with gamma-H2A.1 in ionizing radiation-induced foci (IRIF). Mutational analysis in vitro confirms the functional role of key residues and allows the generation of mutants in which dimerization and phosphopeptide binding are separately disrupted. Phenotypic analysis of these in vivo reveals distinct roles in the DNA damage response. Dimerization mutants are genotoxin sensitive and defective in checkpoint signaling, Chk1 phosphorylation, and Crb2 IRIF formation, while phosphopeptide-binding mutants are only slightly sensitive to IR, have extended checkpoint delays, phosphorylate Chk1, and form Crb2 IRIF. However, disrupting phosphopeptide binding slows formation of ssDNA-binding protein (Rpa1/Rad11) foci and reduces levels of Rad22(Rad52) recombination foci, indicating a DNA repair defect.
Figures







Similar articles
-
BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint.Mol Cell Biol. 2010 Oct;30(19):4732-43. doi: 10.1128/MCB.00413-10. Epub 2010 Aug 2. Mol Cell Biol. 2010. PMID: 20679485 Free PMC article.
-
Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks.Genes Dev. 2006 Jun 15;20(12):1583-96. doi: 10.1101/gad.1422606. Genes Dev. 2006. PMID: 16778077 Free PMC article.
-
Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast.Mol Cell Biol. 2004 Jul;24(14):6215-30. doi: 10.1128/MCB.24.14.6215-6230.2004. Mol Cell Biol. 2004. PMID: 15226425 Free PMC article.
-
Establishment of and recovery from damage checkpoint requires sequential interactions of Crb2 with protein kinases Rad3, Chk1, and Cdc2.Cold Spring Harb Symp Quant Biol. 2000;65:443-9. doi: 10.1101/sqb.2000.65.443. Cold Spring Harb Symp Quant Biol. 2000. PMID: 12760060 Review. No abstract available.
-
14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response.DNA Repair (Amst). 2009 Sep 2;8(9):1009-17. doi: 10.1016/j.dnarep.2009.04.004. Epub 2009 May 29. DNA Repair (Amst). 2009. PMID: 19481982 Free PMC article. Review.
Cited by
-
Requirement for the phospho-H2AX binding module of Crb2 in double-strand break targeting and checkpoint activation.Mol Cell Biol. 2010 Oct;30(19):4722-31. doi: 10.1128/MCB.00404-10. Epub 2010 Aug 2. Mol Cell Biol. 2010. PMID: 20679488 Free PMC article.
-
Eukaryotic DNA damage checkpoint activation in response to double-strand breaks.Cell Mol Life Sci. 2012 May;69(9):1447-73. doi: 10.1007/s00018-011-0875-3. Epub 2011 Nov 15. Cell Mol Life Sci. 2012. PMID: 22083606 Free PMC article. Review.
-
Cardiac and Skeletal Muscle Transcriptome Response to Heat Stress in Kenyan Chicken Ecotypes Adapted to Low and High Altitudes Reveal Differences in Thermal Tolerance and Stress Response.Front Genet. 2019 Oct 11;10:993. doi: 10.3389/fgene.2019.00993. eCollection 2019. Front Genet. 2019. PMID: 31681427 Free PMC article.
-
Critical Function of γH2A in S-Phase.PLoS Genet. 2015 Sep 14;11(9):e1005517. doi: 10.1371/journal.pgen.1005517. eCollection 2015 Sep. PLoS Genet. 2015. PMID: 26368543 Free PMC article.
-
Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications.Genes Dev. 2011 Mar 1;25(5):409-33. doi: 10.1101/gad.2021311. Genes Dev. 2011. PMID: 21363960 Free PMC article. Review.
References
-
- Adams P.D., Gopal K., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Pai R.K., Read R.J., Romo T.D., et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 2004;11:53–55. - PubMed
-
- Ahn J., Urist M., Prives C. Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J. Biol. Chem. 2003;278:20480–20489. - PubMed
-
- Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. - PubMed
-
- Bartek J., Lukas J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr. Opin. Cell Biol. 2001;13:738–747. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous