

The epidermal growth factor homology domain of the LDL receptor drives lipoprotein release through an allosteric mechanism involving H190, H562, and H586
- PMID: 18677035
- PMCID: PMC2546563
- DOI: 10.1074/jbc.M804624200
The epidermal growth factor homology domain of the LDL receptor drives lipoprotein release through an allosteric mechanism involving H190, H562, and H586
Abstract
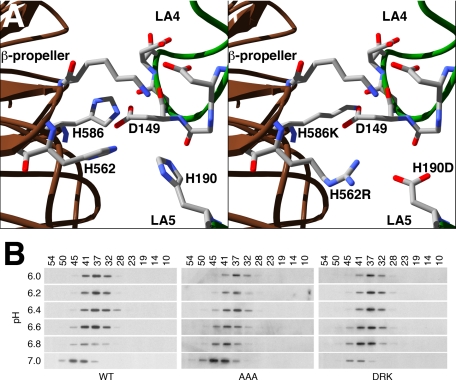
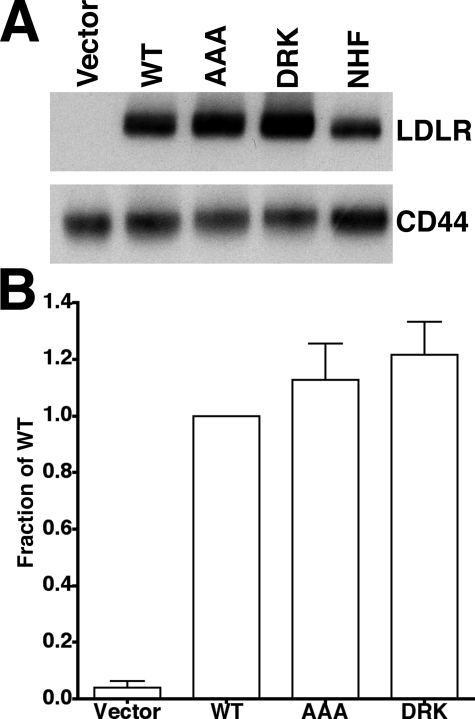
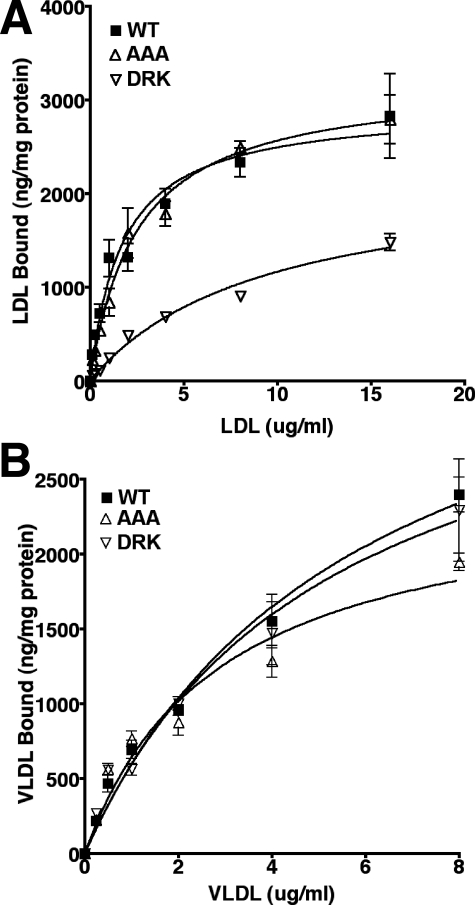
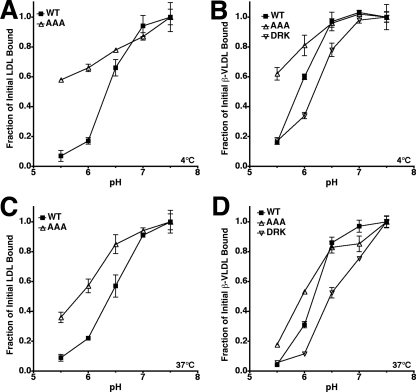
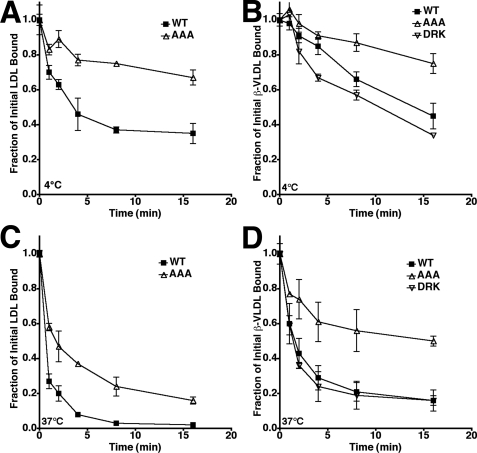
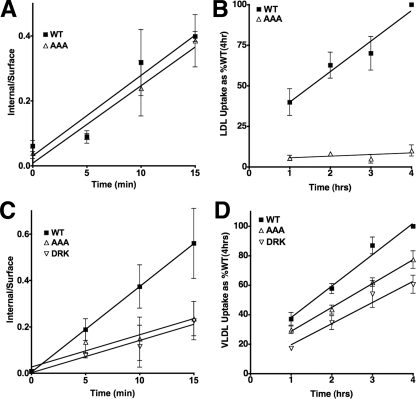
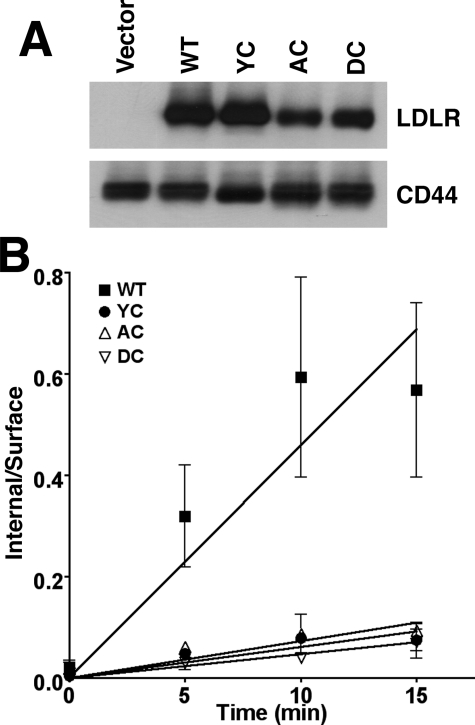
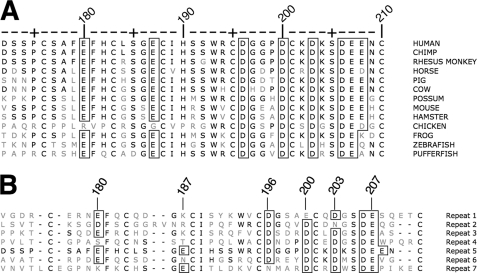
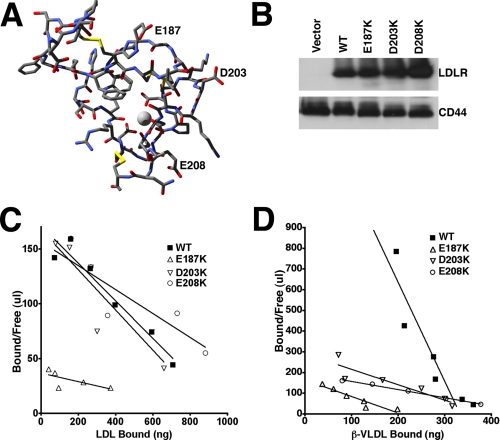
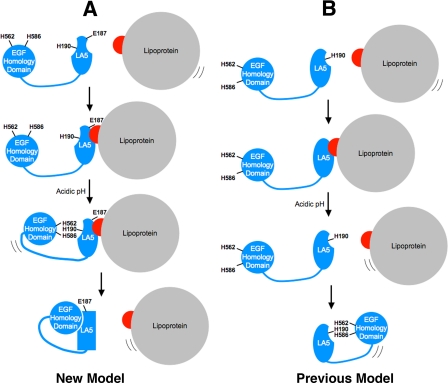
The low density lipoprotein (LDL) receptor (LDLR) mediates efficient endocytosis of VLDL, VLDL remnants, and LDL. As part of the endocytic process, the LDLR releases lipoproteins in endosomes. The release process correlates with an acid-dependent conformational change in the receptor from an extended, "open" state to a compact, "closed" state. The closed state has an intramolecular contact involving H190, H562, and H586. The current model for lipoprotein release holds that protonation of these histidines drives the conformational change that is associated with release. We tested the roles of H190, H562, and H586 on LDLR conformation and on lipoprotein binding, uptake, and release using variants in which the three histidines were replaced with alanine (AAA variant) or in which the histidines were replaced with charged residues that can form ionic contacts at neutral pH (DRK variant). Contrary to expectation, both the AAA and the DRK variants exhibited normal acid-dependent transitions from open to closed conformations. Despite this similarity, both the AAA and DRK mutations modulated lipoprotein release, indicating that H190, H562, and H586 act subsequent to the conformational transition. These observations also suggest that the intramolecular contact does not drive release through a competitive mechanism. In support of this possibility, mutagenesis experiments showed that beta-VLDL binding was inhibited by mutations at D203 and E208, which are exposed in the closed conformation of the LDLR. We propose that H190, H562, and H586 are part of an allosteric mechanism that drives lipoprotein release.
Figures
Similar articles
-
The role of calcium in lipoprotein release by the low-density lipoprotein receptor.Biochemistry. 2009 Aug 4;48(30):7313-24. doi: 10.1021/bi900214u. Biochemistry. 2009. PMID: 19583244 Free PMC article.
-
Quantitative fluorescence imaging reveals point of release for lipoproteins during LDLR-dependent uptake.J Lipid Res. 2013 Mar;54(3):744-753. doi: 10.1194/jlr.M033548. Epub 2013 Jan 7. J Lipid Res. 2013. PMID: 23296879 Free PMC article.
-
Role of an intramolecular contact on lipoprotein uptake by the LDL receptor.Biochim Biophys Acta. 2011 Jun;1811(6):397-408. doi: 10.1016/j.bbalip.2011.04.002. Epub 2011 Apr 9. Biochim Biophys Acta. 2011. PMID: 21511053 Free PMC article.
-
The LDL receptor: how acid pulls the trigger.Trends Biochem Sci. 2005 Jun;30(6):309-17. doi: 10.1016/j.tibs.2005.03.007. Trends Biochem Sci. 2005. PMID: 15950875 Review.
-
The low-density lipoprotein receptor: ligands, debates and lore.Curr Opin Struct Biol. 2003 Dec;13(6):683-9. doi: 10.1016/j.sbi.2003.10.001. Curr Opin Struct Biol. 2003. PMID: 14675545 Review.
Cited by
-
Model of human low-density lipoprotein and bound receptor based on cryoEM.Proc Natl Acad Sci U S A. 2010 Jan 19;107(3):1059-64. doi: 10.1073/pnas.0908004107. Epub 2009 Dec 28. Proc Natl Acad Sci U S A. 2010. PMID: 20080547 Free PMC article.
-
Domain swapping reveals that low density lipoprotein (LDL) type A repeat order affects ligand binding to the LDL receptor.J Biol Chem. 2009 May 15;284(20):13396-13400. doi: 10.1074/jbc.M900194200. Epub 2009 Mar 26. J Biol Chem. 2009. PMID: 19329437 Free PMC article.
-
The role of calcium in lipoprotein release by the low-density lipoprotein receptor.Biochemistry. 2009 Aug 4;48(30):7313-24. doi: 10.1021/bi900214u. Biochemistry. 2009. PMID: 19583244 Free PMC article.
-
Identification of roles for H264, H306, H439, and H635 in acid-dependent lipoprotein release by the LDL receptor.J Lipid Res. 2017 Feb;58(2):364-374. doi: 10.1194/jlr.M070938. Epub 2016 Nov 28. J Lipid Res. 2017. PMID: 27895090 Free PMC article.
-
Endocrine Regulation of Microvascular Receptor-Mediated Transcytosis and Its Therapeutic Opportunities: Insights by PCSK9-Mediated Regulation.Pharmaceutics. 2023 Apr 18;15(4):1268. doi: 10.3390/pharmaceutics15041268. Pharmaceutics. 2023. PMID: 37111752 Free PMC article. Review.
References
-
- Yamamoto, T., Davis, C. G., Brown, M. S., Schneider, W. J., Casey, M. L., Goldstein, J. L., and Russell, D. W. (1984) Cell 3927 -38 - PubMed
-
- Esser, V., Limbird, L. E., Brown, M. S., Goldstein, J. L., and Russell, D. W. (1988) J. Biol. Chem. 26313282 -13290 - PubMed
-
- North, C. L., and Blacklow, S. C. (2000) Biochemistry 392564 -2571 - PubMed
-
- Davis, C. G., Goldstein, J. L., Sudhof, T. C., Anderson, R. G., Russell, D. W., and Brown, M. S. (1987) Nature 326760 -765 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
