

Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins
- PMID: 18677408
- PMCID: PMC2491457
- DOI: 10.1172/JCI34120
Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins
Abstract
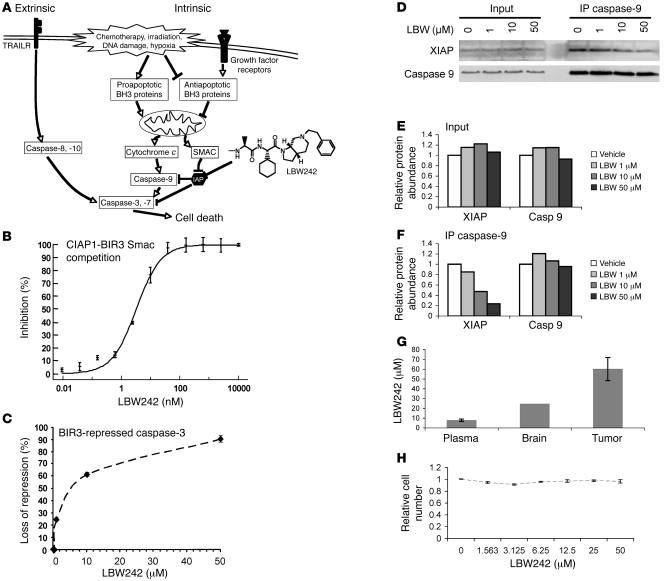
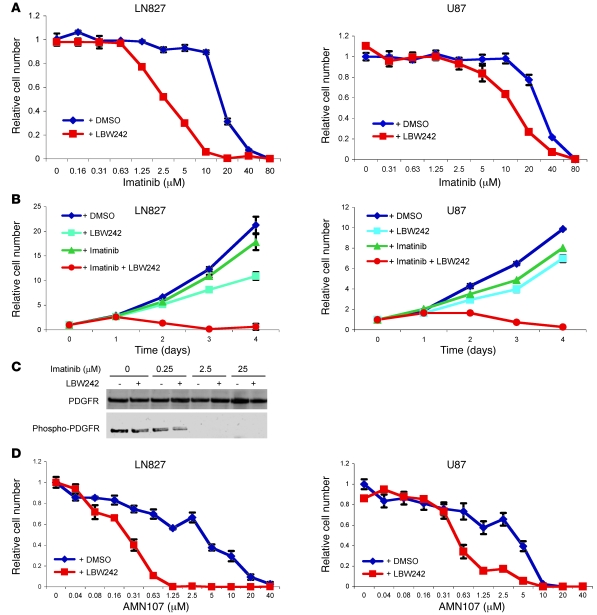
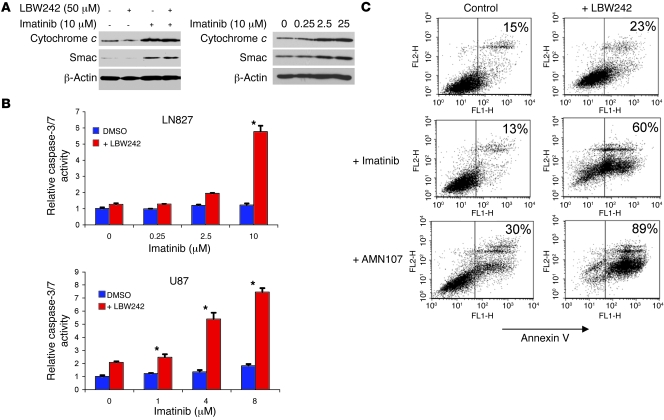
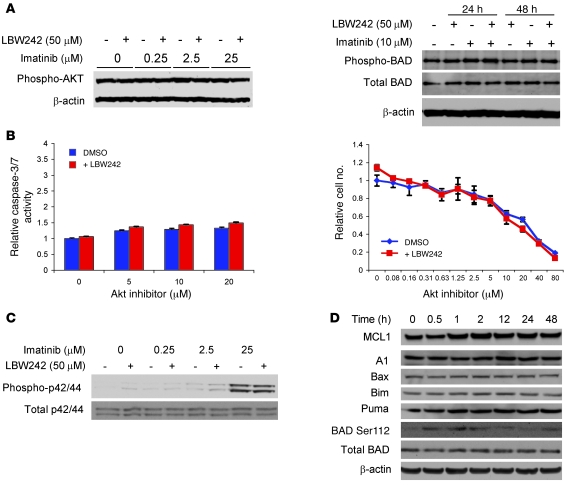
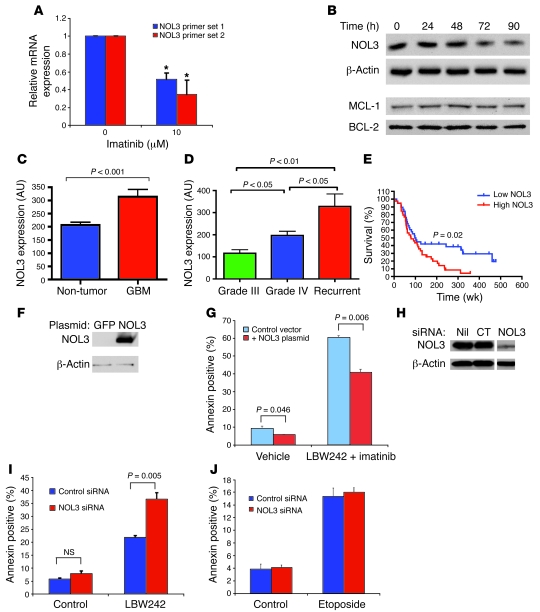
Multiple receptor tyrosine kinases (RTKs), including PDGFR, have been validated as therapeutic targets in glioblastoma multiforme (GBM), yet inhibitors of RTKs have had limited clinical success. As various antiapoptotic mechanisms render GBM cells resistant to chemo- and radiotherapy, we hypothesized that these antiapoptotic mechanisms also confer resistance to RTK inhibition. We found that in vitro inhibition of PDGFR in human GBM cells initiated the intrinsic pathway of apoptosis, as evidenced by mitochondrial outer membrane permeabilization, but downstream caspase activation was blocked by inhibitor of apoptosis proteins (IAPs). Consistent with this, inhibition of PDGFR combined with small molecule inactivation of IAPs induced apoptosis in human GBM cells in vitro and had synergistic antitumor effects in orthotopic mouse models of GBM and in primary human GBM neurospheres. These results demonstrate that concomitant inhibition of IAPs can overcome resistance to RTK inhibitors in human malignant GBM cells, and suggest that blockade of IAPs has the potential to improve treatment outcomes in patients with GBM.
Figures

References
-
- Gammeltoft S., Ballotti R., Kowalski A., Westermark B., Van Obberghen E. Expression of two types of receptor for insulin-like growth factors in human malignant glioma. Cancer Res. 1988;48:1233–1237. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
