

Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia
- PMID: 18677410
- PMCID: PMC2491459
- DOI: 10.1172/JCI35090
Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia
Abstract
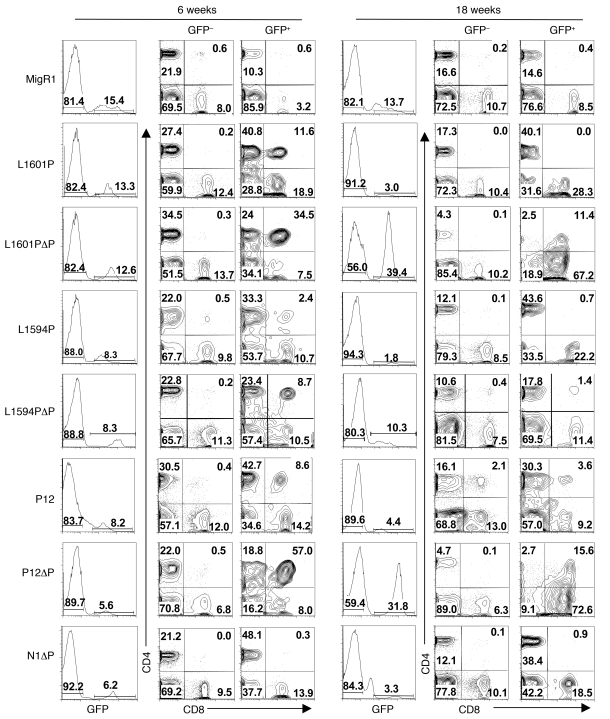
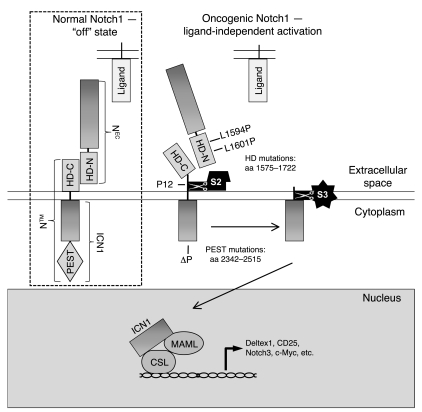
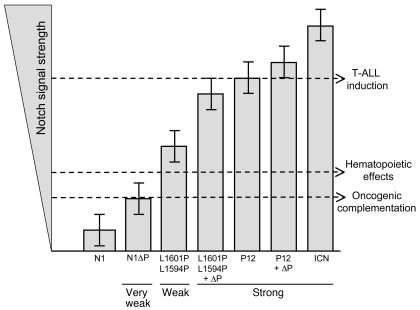
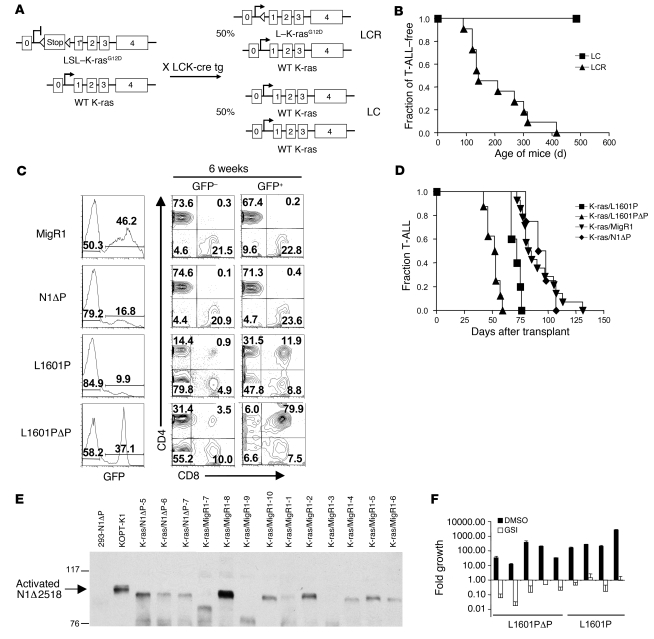
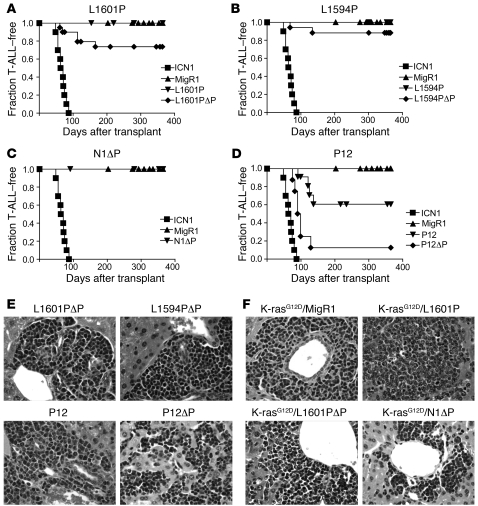
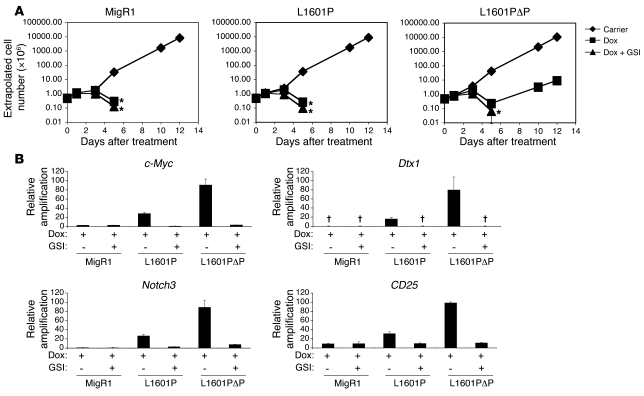
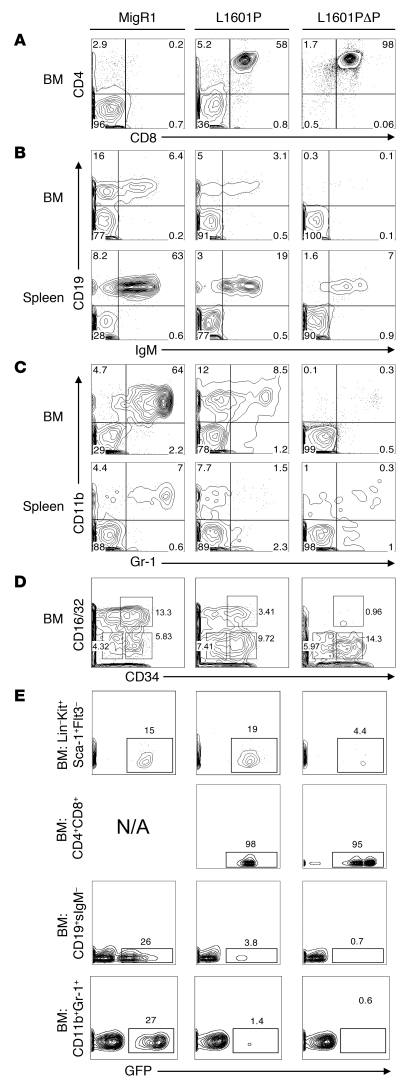
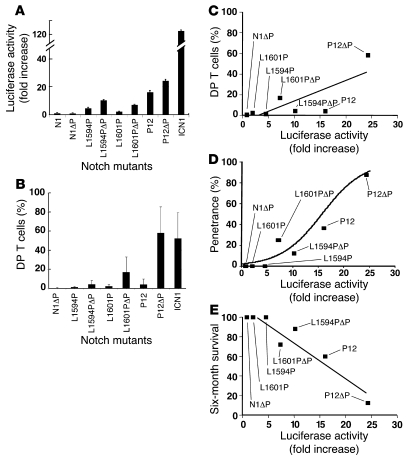
Gain-of-function NOTCH1 mutations are found in 50%-70% of human T cell acute lymphoblastic leukemia/lymphoma (T-ALL) cases. Gain-of-function NOTCH1 alleles that initiate strong downstream signals induce leukemia in mice, but it is unknown whether the gain-of-function NOTCH1 mutations most commonly found in individuals with T-ALL generate downstream signals of sufficient strength to induce leukemia. We addressed this question by expressing human gain-of-function NOTCH1 alleles of varying strength in mouse hematopoietic precursors. Uncommon gain-of-function NOTCH1 alleles that initiated strong downstream signals drove ectopic T cell development and induced leukemia efficiently. In contrast, although gain-of-function alleles that initiated only weak downstream signals also induced ectopic T cell development, these more common alleles failed to efficiently initiate leukemia development. However, weak gain-of-function NOTCH1 alleles accelerated the onset of leukemia initiated by constitutively active K-ras and gave rise to tumors that were sensitive to Notch signaling pathway inhibition. These data show that induction of leukemia requires doses of Notch1 greater than those needed for T cell development and that most NOTCH1 mutations found in T-ALL cells do not generate signals of sufficient strength to initiate leukemia development. Furthermore, low, nonleukemogenic levels of Notch1 can complement other leukemogenic events, such as activation of K-ras. Even when Notch1 participates secondarily, the resulting tumors show "addiction" to Notch, providing a further rationale for evaluating Notch signaling pathway inhibitors in leukemia.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
