

PINK1 mutations and parkinsonism
- PMID: 18685134
- PMCID: PMC2676945
- DOI: 10.1212/01.wnl.0000323812.40708.1f
PINK1 mutations and parkinsonism
Abstract
Background: PINK1 loss-of-function causes recessive, early-onset parkinsonism. In Tunisia there is a high rate of consanguineous marriage but PINK1 carrier frequency and disease prevalence have yet to be assessed.
Objectives: The frequency of PINK1 mutations in familial parkinsonism, community-based patients with idiopathic Parkinson disease (PD) (non-familial PD), and control subjects was determined. Demographic and clinical characteristics of individuals with PINK1 homozygous or heterozygous variants, or without PINK1 mutations, were compared.
Methods: A total of 92 kindreds (with 208 affected and 340 unaffected subjects), 240 nonfamilial PD, and 368 control participants were recruited from the Institut National de Neurologie, Tunis. Clinical examinations included Hoehn &Yahr, UPDRS, and Epworth scales. PINK1 sequencing and dosage analysis was performed in familial index patients, the variants identified screened in all subjects. Parkin and LRRK2 genes were also examined.
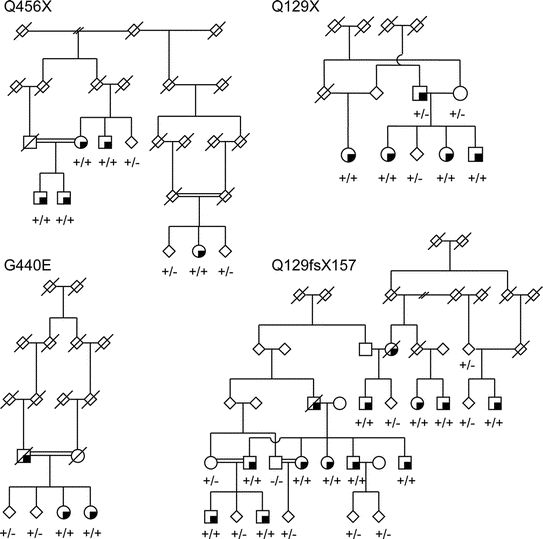
Results: Four PINK1 homozygous mutations, three novel (Q129X, Q129fsX157, G440E, and one previously reported; Q456X), segregate with parkinsonism in 46 individuals in 14 of 92 families (15%). Six of 240 patients with nonfamilial PD were found with either homozygous Q456X or Q129X (2.5%) substitutions. In patients with familial disease, PINK1 homozygotes were younger at disease onset (36 +/- 12 years) than noncarriers (57 +/- 15 years) and more often had an akinetic-rigid presentation at examination and slow progression.
Conclusions: Segregation of PINK1 mutations with parkinsonism within families, and frequency estimates within population controls, suggested only four PINK1 mutations were pathogenic. Several PINK1 sequence variants are potentially benign and there was no evidence that PINK1 heterozygosity increases susceptibility to idiopathic Parkinson disease.
Figures
References
-
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 2006;7:306–318. - PubMed
-
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160. - PubMed
-
- Silvestri L, Caputo V, Bellacchio E, et al. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 2005;14:3477–3492. - PubMed
-
- Gouider-Khouja N, Belal S, Hamida MB, Hentati F. Clinical and genetic study of familial Parkinson's disease in Tunisia. Neurology 2000;54:1603–1609. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources