

Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels
- PMID: 18687887
- PMCID: PMC2503926
- DOI: 10.1073/pnas.0804350105
Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels
Abstract
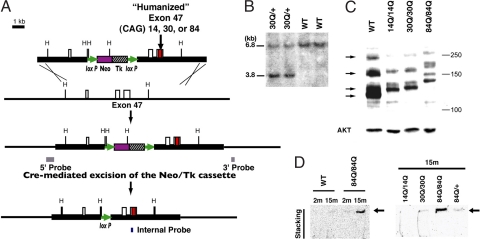
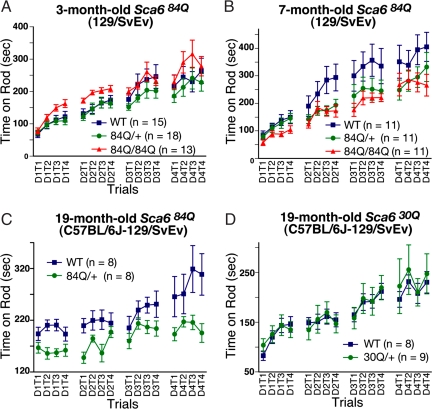
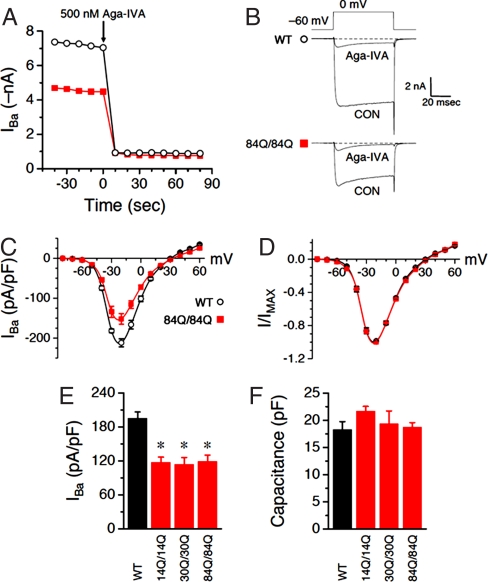
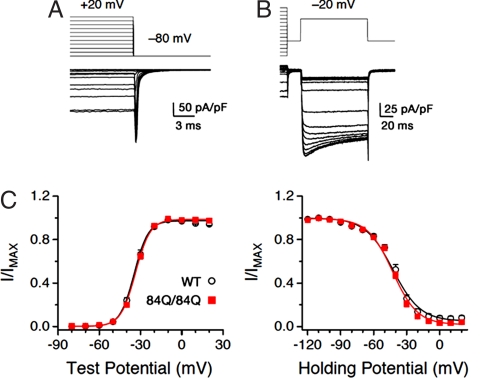
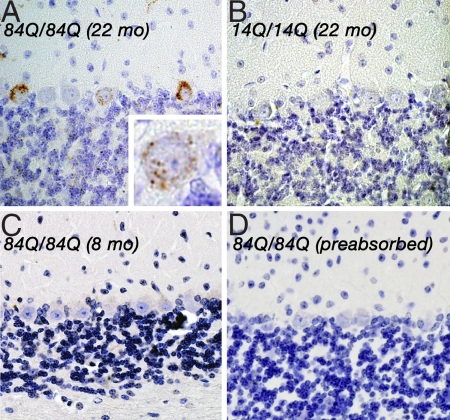
Spinocerebellar ataxia type 6 (SCA6) is a neurodegenerative disorder caused by CAG repeat expansions within the voltage-gated calcium (Ca(V)) 2.1 channel gene. It remains controversial whether the mutation exerts neurotoxicity by changing the function of Ca(V)2.1 channel or through a gain-of-function mechanism associated with accumulation of the expanded polyglutamine protein. We generated three strains of knockin (KI) mice carrying normal, expanded, or hyperexpanded CAG repeat tracts in the Cacna1a locus. The mice expressing hyperexpanded polyglutamine (Sca6(84Q)) developed progressive motor impairment and aggregation of mutant Ca(V)2.1 channels. Electrophysiological analysis of cerebellar Purkinje cells revealed similar Ca(2+) channel current density among the three KI models. Neither voltage sensitivity of activation nor inactivation was altered in the Sca6(84Q) neurons, suggesting that expanded CAG repeat per se does not affect the intrinsic electrophysiological properties of the channels. The pathogenesis of SCA6 is apparently linked to an age-dependent process accompanied by accumulation of mutant Ca(V)2.1 channels.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Zhuchenko O, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage dependent calcium channel. Nat Genet. 1997;15:62–69. - PubMed
-
- Tsuchiya K, et al. Degeneration of the inferior olive in spinocerebellar ataxia 6 may depend on disease duration: Report of two autopsy cases and statistical analysis of autopsy cases reported to date. Neuropathology. 2005;25:125–135. - PubMed
-
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. - PubMed
-
- Mariotti C, et al. Pathogenic effect of an intermediate-size SCA6-allele (CAG)(19) in a homozygous patient. Neurology. 2001;57:1502–1504. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
