

Comparison of the Agilent, ROMA/NimbleGen and Illumina platforms for classification of copy number alterations in human breast tumors
- PMID: 18691401
- PMCID: PMC2547478
- DOI: 10.1186/1471-2164-9-379
Comparison of the Agilent, ROMA/NimbleGen and Illumina platforms for classification of copy number alterations in human breast tumors
Abstract
Background: Microarray Comparative Genomic Hybridization (array CGH) provides a means to examine DNA copy number aberrations. Various platforms, brands and underlying technologies are available, facing the user with many choices regarding platform sensitivity and number, localization, and density distribution of probes.
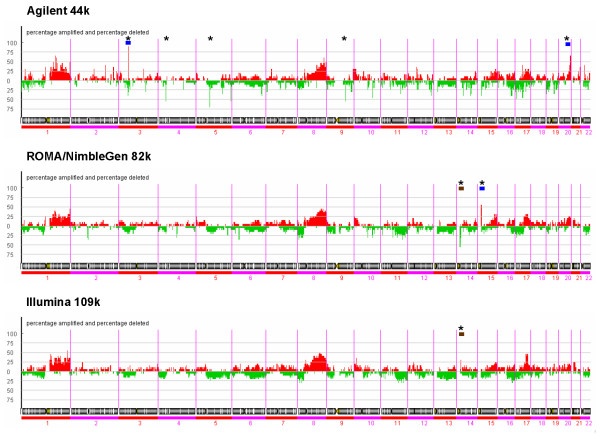
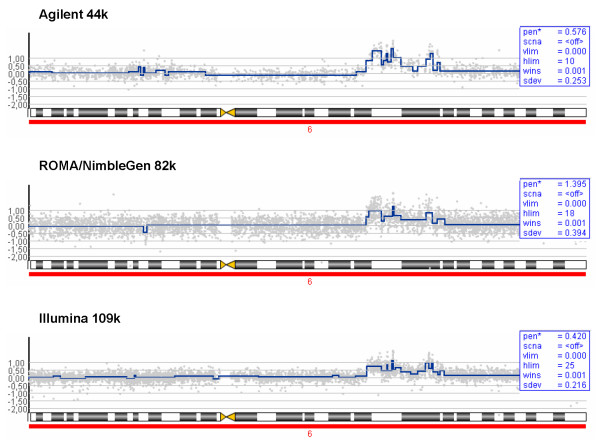
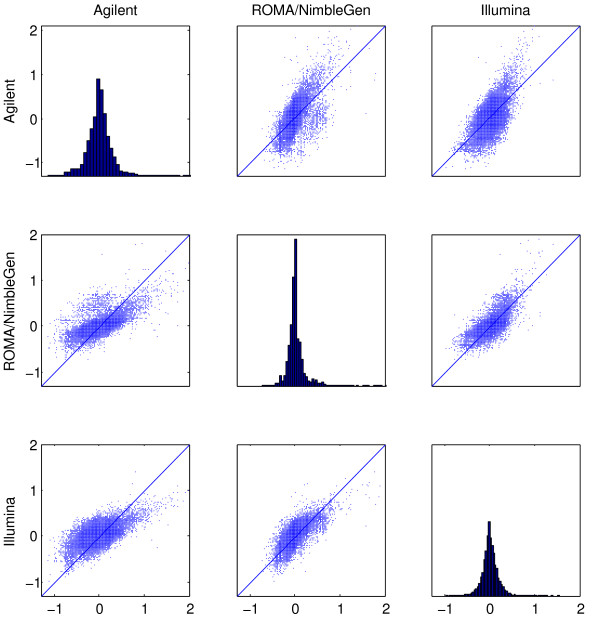
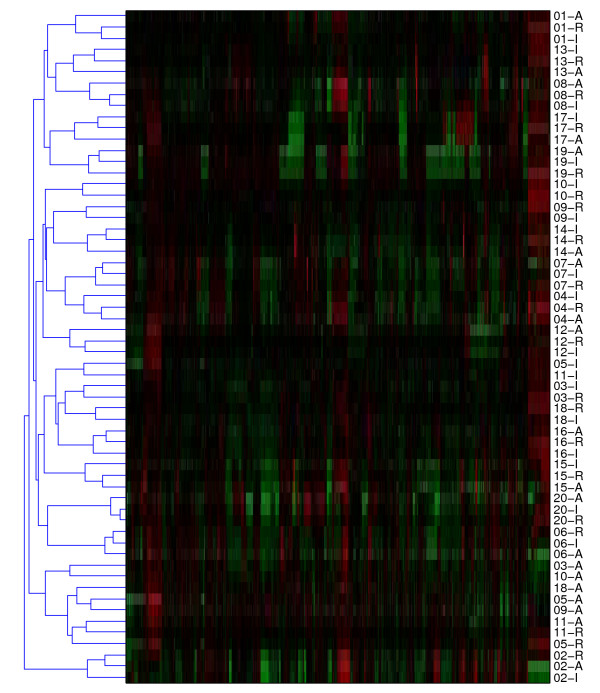
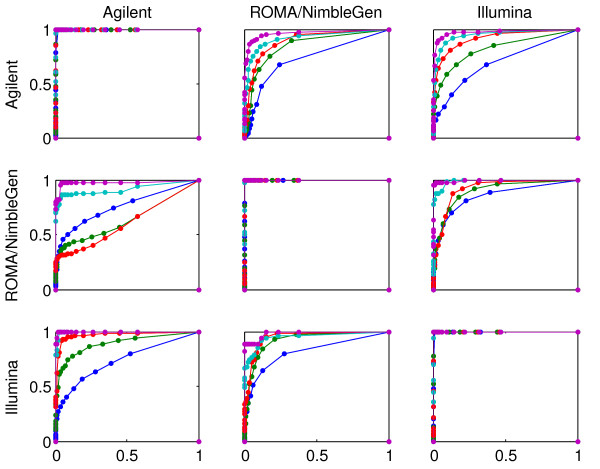
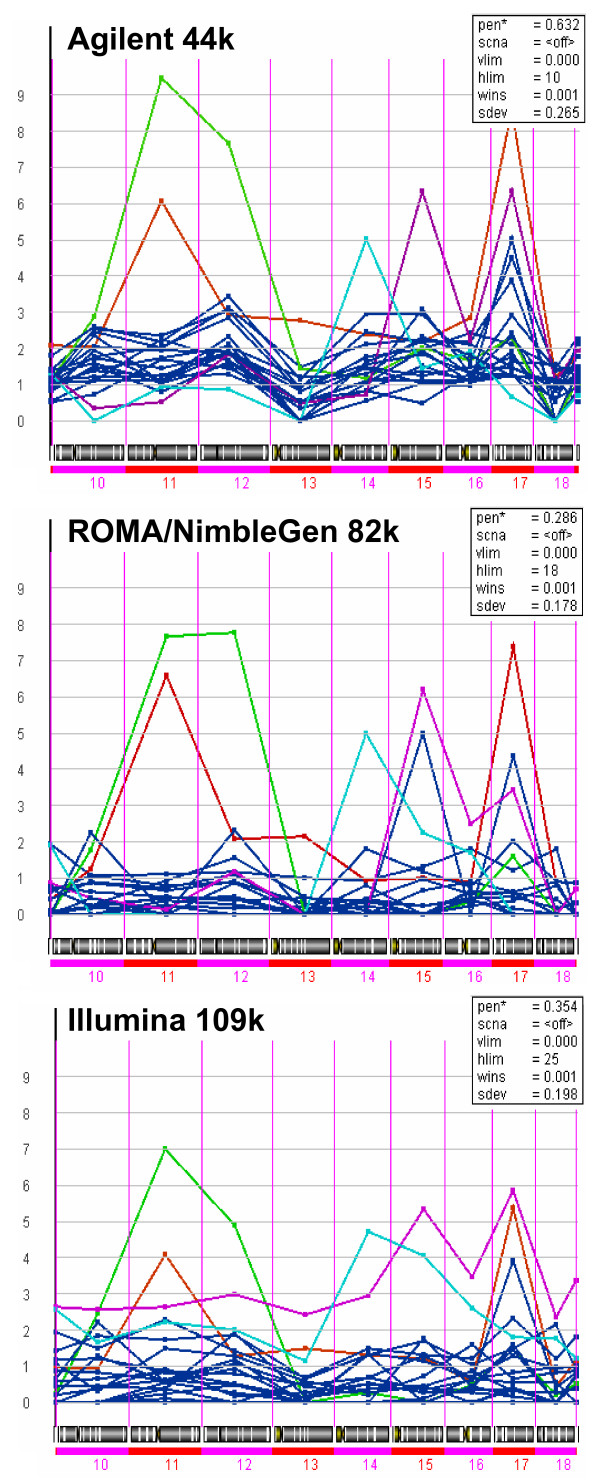
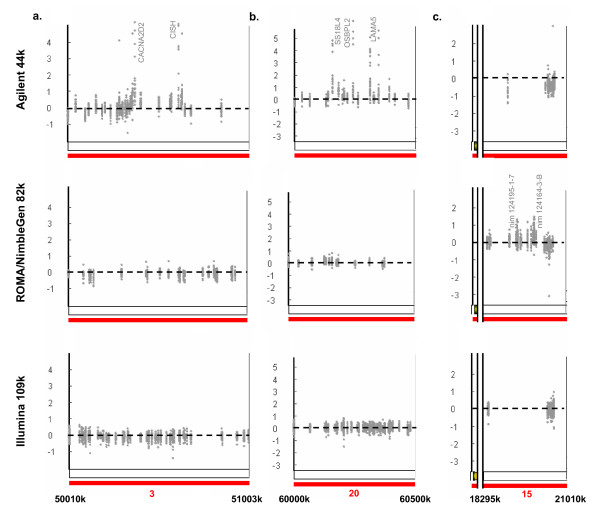
Results: We evaluate three different platforms presenting different nature and arrangement of the probes: The Agilent Human Genome CGH Microarray 44 k, the ROMA/NimbleGen Representational Oligonucleotide Microarray 82 k, and the Illumina Human-1 Genotyping 109 k BeadChip, with Agilent being gene oriented, ROMA/NimbleGen being genome oriented, and Illumina being genotyping oriented. We investigated copy number changes in 20 human breast tumor samples representing different gene expression subclasses, using a suite of graphical and statistical methods designed to work across platforms. Despite substantial differences in the composition and spatial distribution of probes, the comparison revealed high overall concordance. Notably however, some short amplifications and deletions of potential biological importance were not detected by all platforms. Both correlation and cluster analysis indicate a somewhat higher similarity between ROMA/NimbleGen and Illumina than between Agilent and the other two platforms. The programs developed for the analysis are available from http://www.ifi.uio.no/bioinf/Projects/.
Conclusion: We conclude that platforms based on different technology principles reveal similar aberration patterns, although we observed some unique amplification or deletion peaks at various locations, only detected by one of the platforms. The correct platform choice for a particular study is dependent on whether the appointed research intention is gene, genome, or genotype oriented.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
