

Disease-specific induced pluripotent stem cells
- PMID: 18691744
- PMCID: PMC2633781
- DOI: 10.1016/j.cell.2008.07.041
Disease-specific induced pluripotent stem cells
Abstract
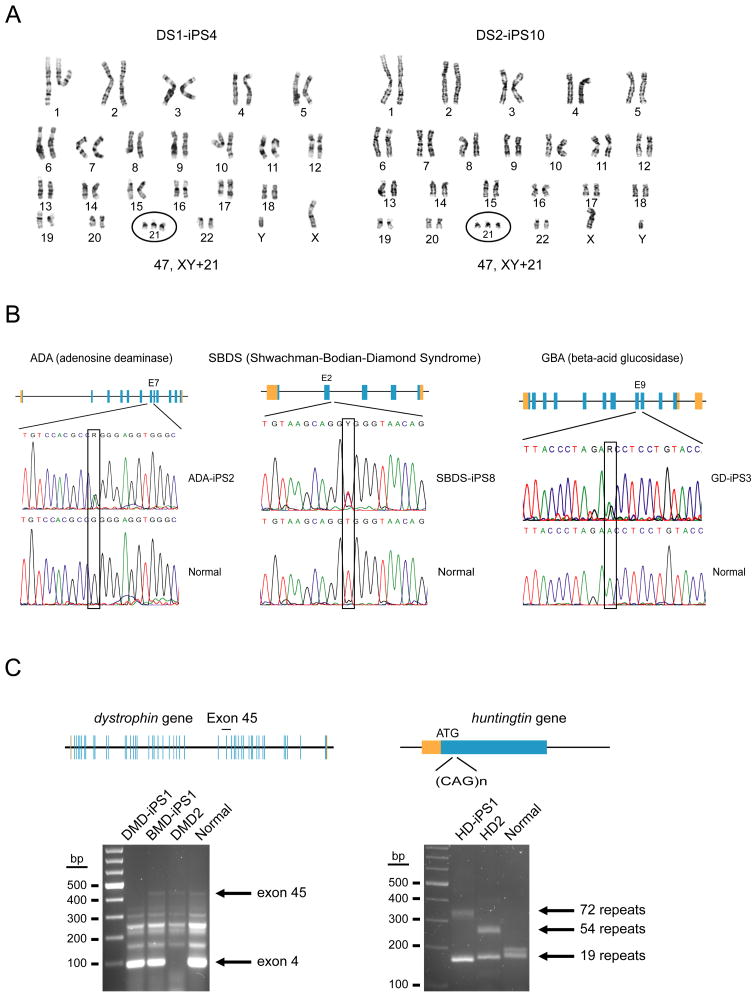
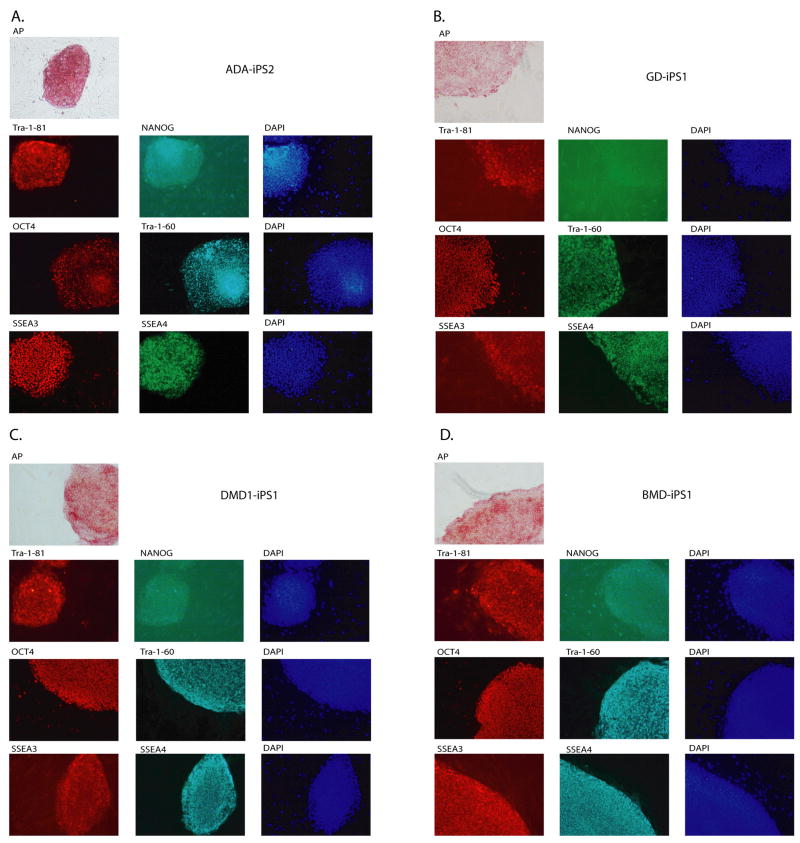
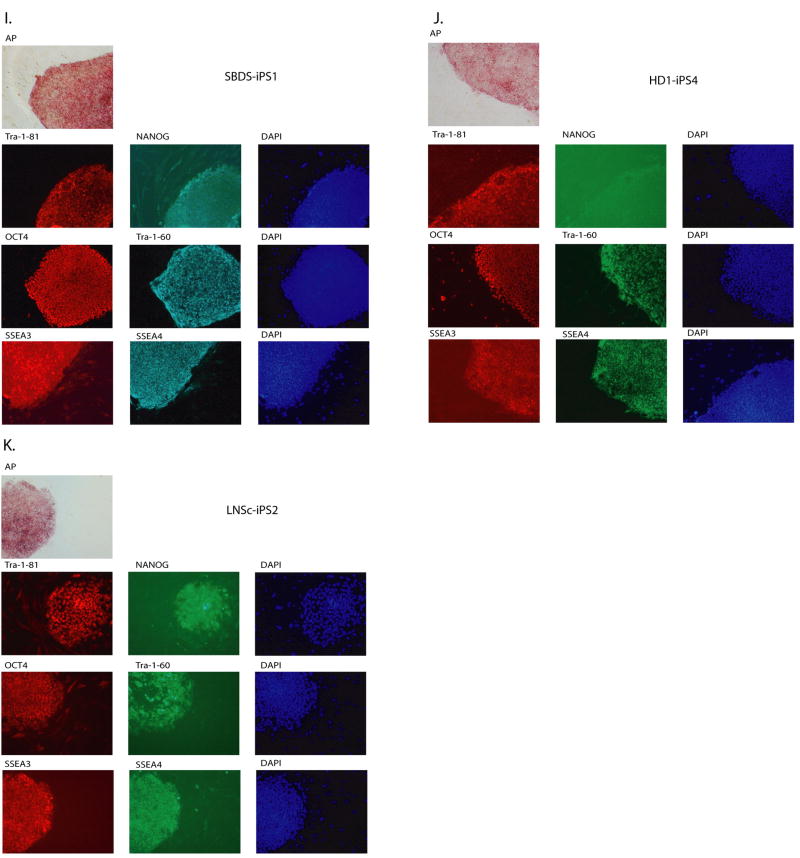
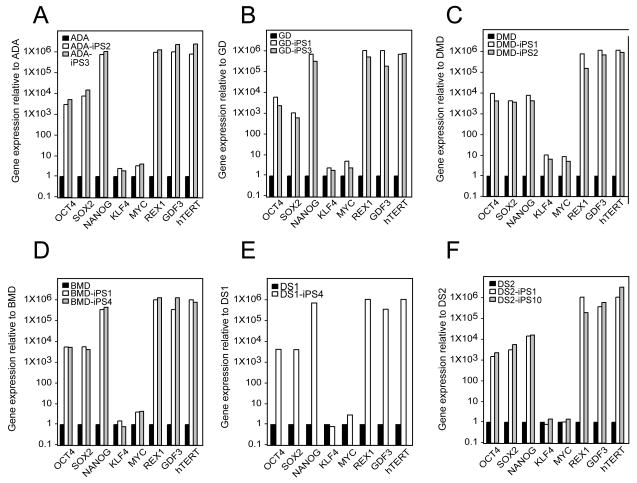
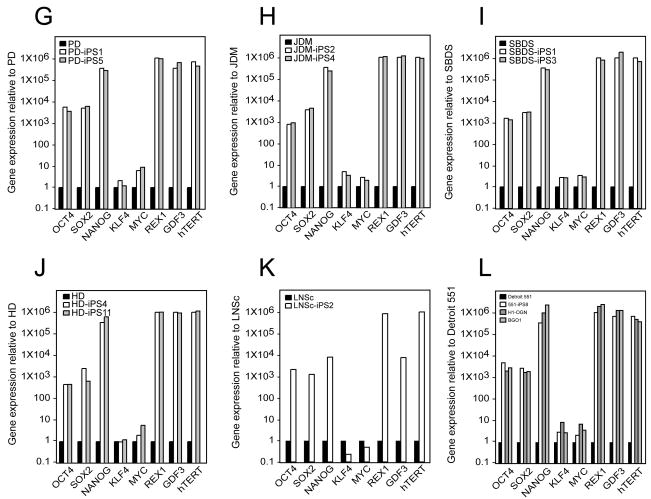
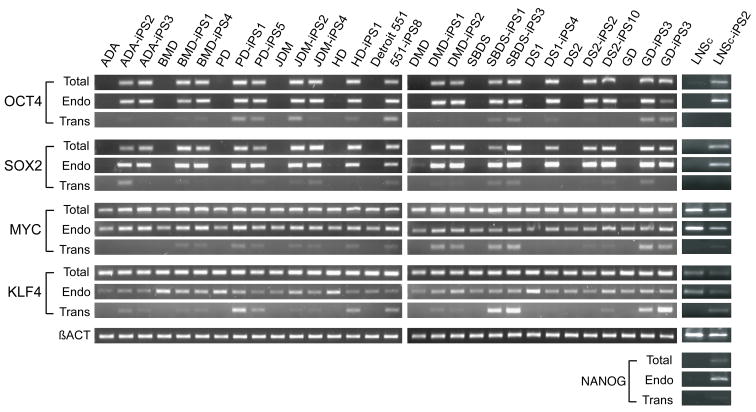
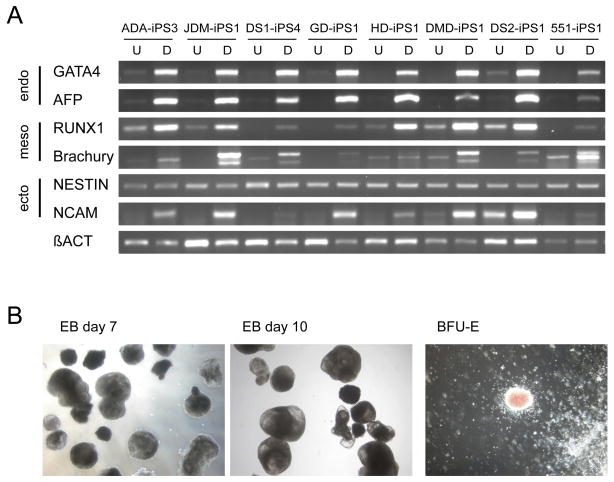
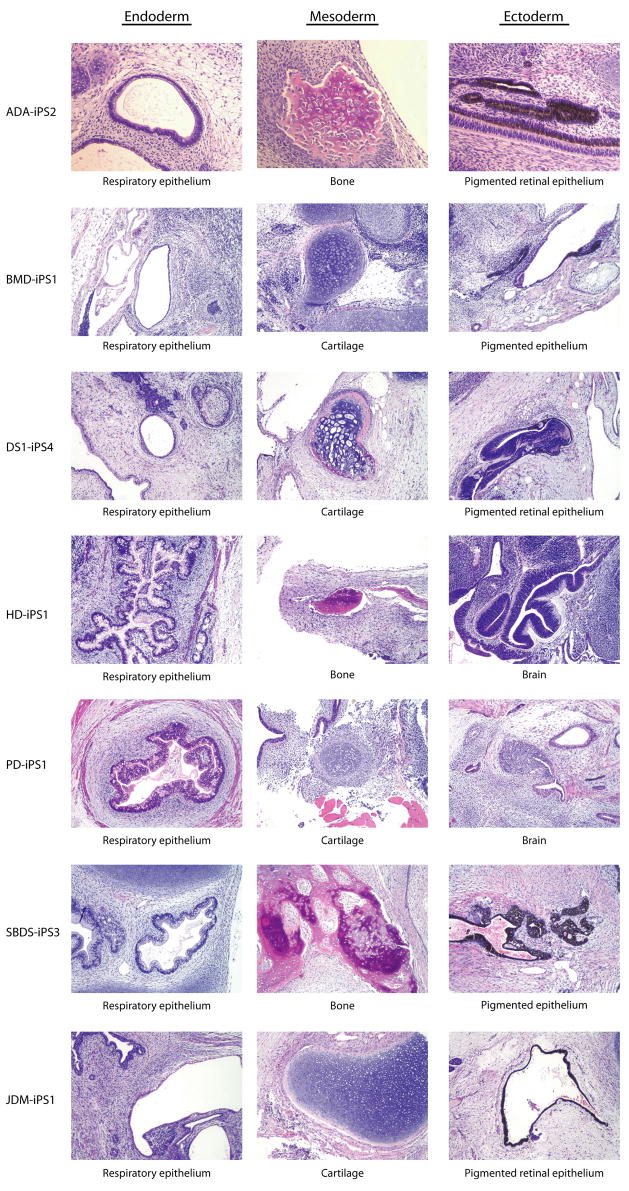
Tissue culture of immortal cell strains from diseased patients is an invaluable resource for medical research but is largely limited to tumor cell lines or transformed derivatives of native tissues. Here we describe the generation of induced pluripotent stem (iPS) cells from patients with a variety of genetic diseases with either Mendelian or complex inheritance; these diseases include adenosine deaminase deficiency-related severe combined immunodeficiency (ADA-SCID), Shwachman-Bodian-Diamond syndrome (SBDS), Gaucher disease (GD) type III, Duchenne (DMD) and Becker muscular dystrophy (BMD), Parkinson disease (PD), Huntington disease (HD), juvenile-onset, type 1 diabetes mellitus (JDM), Down syndrome (DS)/trisomy 21, and the carrier state of Lesch-Nyhan syndrome. Such disease-specific stem cells offer an unprecedented opportunity to recapitulate both normal and pathologic human tissue formation in vitro, thereby enabling disease investigation and drug development.
Figures

References
-
- Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725–738. - PubMed
-
- Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86:45–48. - PubMed
-
- Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. Eur J Public Health. 2007;17:221–225. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
