

Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells
- PMID: 18694499
- PMCID: PMC2527015
- DOI: 10.1186/1471-2407-8-229
Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells
Abstract
Background: Although lung cancer is among the few malignancies for which we know the primary etiological agent (i.e., cigarette smoke), a precise understanding of the temporal sequence of events that drive tumor progression remains elusive. In addition to finding that cigarette smoke (CS) impacts the functioning of key pathways with significant roles in redox homeostasis, xenobiotic detoxification, cell cycle control, and endoplasmic reticulum (ER) functioning, our data highlighted a defensive role for the unfolded protein response (UPR) program. The UPR promotes cell survival by reducing the accumulation of aberrantly folded proteins through translation arrest, production of chaperone proteins, and increased degradation. Importance of the UPR in maintaining tissue health is evidenced by the fact that a chronic increase in defective protein structures plays a pathogenic role in diabetes, cardiovascular disease, Alzheimer's and Parkinson's syndromes, and cancer.
Methods: Gene and protein expression changes in CS exposed human cell cultures were monitored by high-density microarrays and Western blot analysis. Tissue arrays containing samples from 110 lung cancers were probed with antibodies to proteins of interest using immunohistochemistry.
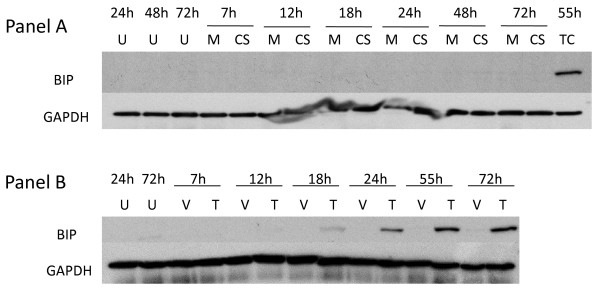
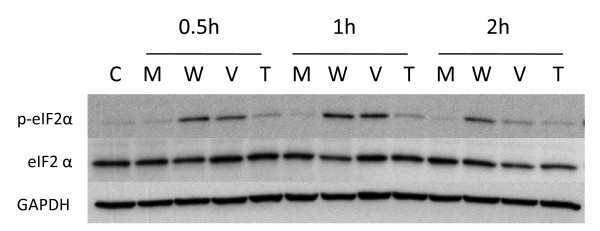
Results: We show that: 1) CS induces ER stress and activates components of the UPR; 2) reactive species in CS that promote oxidative stress are primarily responsible for UPR activation; 3) CS exposure results in increased expression of several genes with significant roles in attenuating oxidative stress; and 4) several major UPR regulators are increased either in expression (i.e., BiP and eIF2 alpha) or phosphorylation (i.e., phospho-eIF2 alpha) in a majority of human lung cancers.
Conclusion: These data indicate that chronic ER stress and recruitment of one or more UPR effector arms upon exposure to CS may play a pivotal role in the etiology or progression of lung cancers, and that phospho-eIF2 alpha and BiP may have diagnostic and/or therapeutic potential. Furthermore, we speculate that upregulation of UPR regulators (in particular BiP) may provide a pro-survival advantage by increasing resistance to cytotoxic stresses such as hypoxia and chemotherapeutic drugs, and that UPR induction is a potential mechanism that could be attenuated or reversed resulting in a more efficacious treatment strategy for lung cancer.
Figures










References
-
- Lubin JH, Alavanja MC, Caporaso N, Brown LM, Brownson RC, Field RW, Garcia-Closas M, Hartge P, Hauptmann M, Hayes RB, Kleinerman R, Kogevinas M, Krewski D, Langholz B, Letourneau EG, Lynch CF, Malats N, Sandler DP, Schaffrath-Rosario A, Schoenberg JB, Silverman DT, Wang Z, Wichmann HE, Wilcox HB, Zielinski JM. Cigarette smoking and cancer risk: modeling total exposure and intensity. Am J Epidemiol. 2007;166:479–489. - PubMed
-
- Alberg AJ, Ford JG, Samet JM. Epidemiology of lung cancer: ACCP evidence-based clinical practice guidelines (2nd edition) Chest. 2007;132:29S–55S. - PubMed
-
- Albino AP, Huang X, Jorgensen E, Yang J, Gietl D, Traganos F, Darzynkiewicz Z. Induction of H2AX phosphorylation in pulmonary cells by tobacco smoke: a new assay for carcinogens. Cell Cycle. 2004;3:1062–1068. - PubMed
-
- Jorgensen ED, Dozmorov I, Frank MB, Centola M, Albino AP. Global Gene Expression Analysis of Human Bronchial Epithelial Cells Treated with Tobacco Condensates. Cell Cycle. 2004;3:1154–1168. - PubMed
-
- Albino AP, Huang X, Jorgensen ED, Gietl D, Traganos F, Darzynkiewicz Z. Induction of DNA double-strand breaks in A549 and normal human pulmonary epithelial cells by cigarette smoke is mediated by free radicals. Int J Oncol. 2006;28:1491–1505. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
