

The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis
- PMID: 18694565
- PMCID: PMC2685763
- DOI: 10.1016/j.devcel.2008.07.002
The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis
Abstract
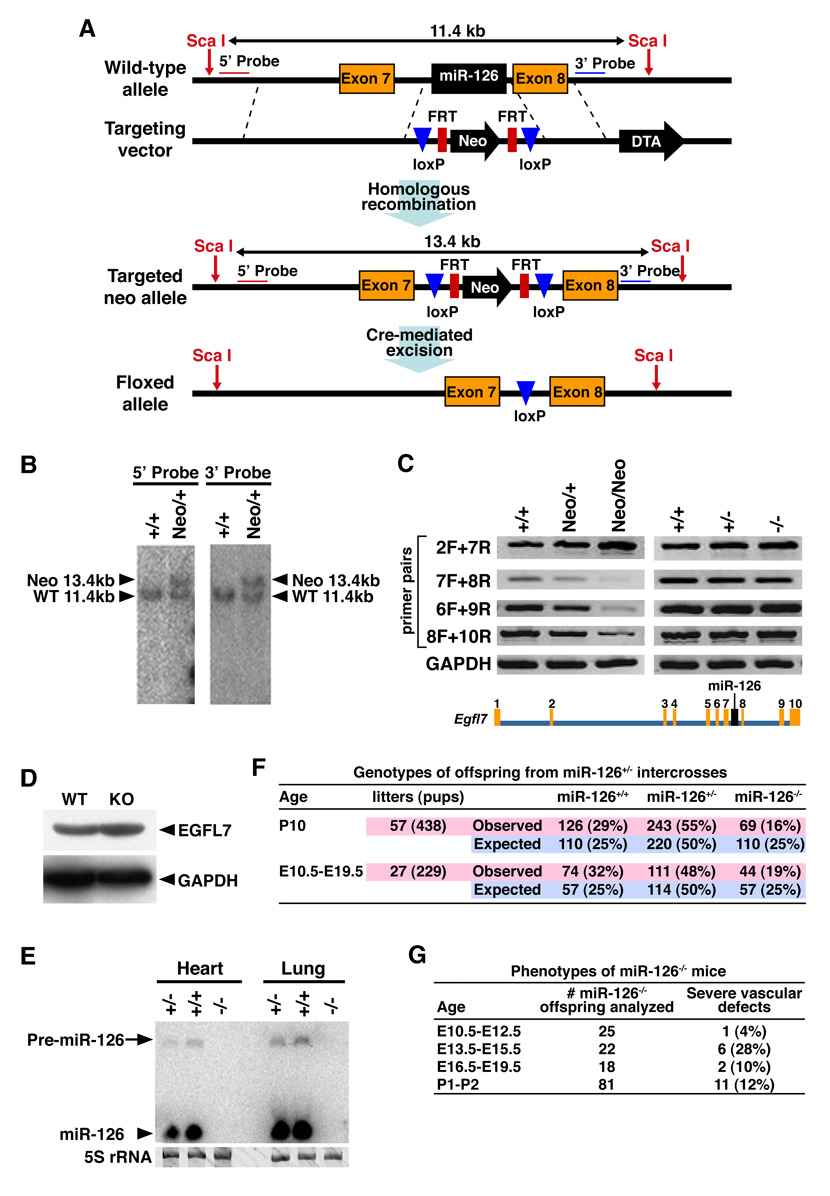
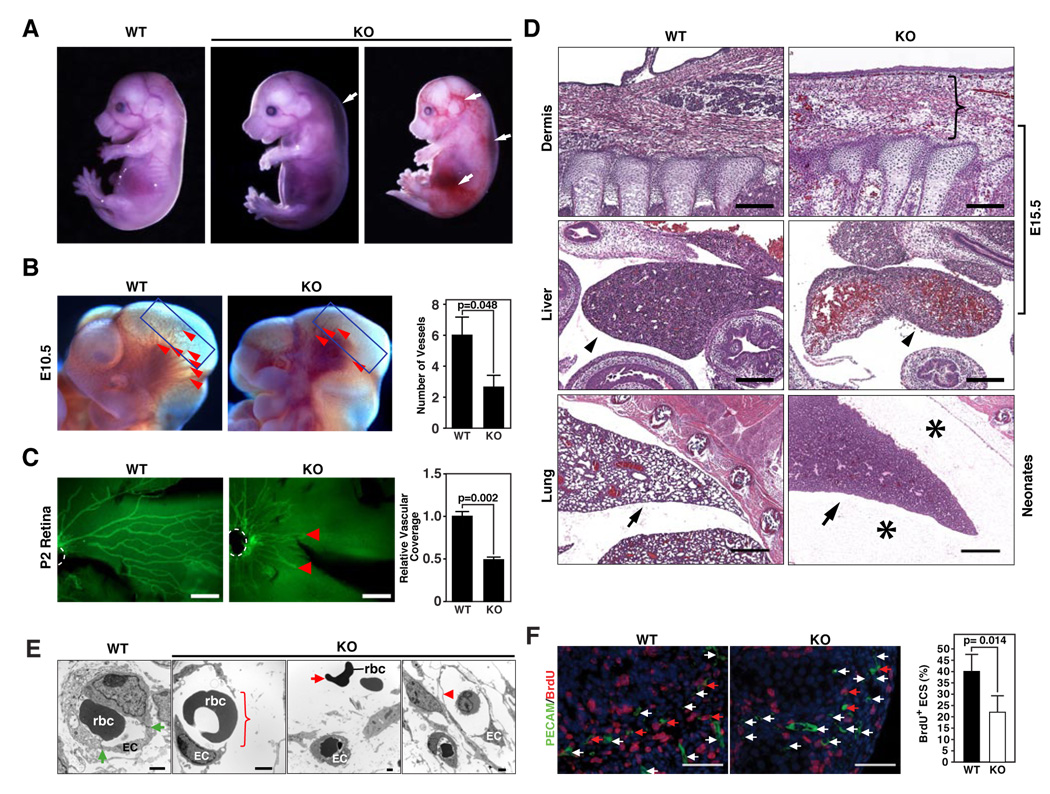
Endothelial cells play essential roles in maintenance of vascular integrity, angiogenesis, and wound repair. We show that an endothelial cell-restricted microRNA (miR-126) mediates developmental angiogenesis in vivo. Targeted deletion of miR-126 in mice causes leaky vessels, hemorrhaging, and partial embryonic lethality, due to a loss of vascular integrity and defects in endothelial cell proliferation, migration, and angiogenesis. The subset of mutant animals that survives displays defective cardiac neovascularization following myocardial infarction. The vascular abnormalities of miR-126 mutant mice resemble the consequences of diminished signaling by angiogenic growth factors, such as VEGF and FGF. Accordingly, miR-126 enhances the proangiogenic actions of VEGF and FGF and promotes blood vessel formation by repressing the expression of Spred-1, an intracellular inhibitor of angiogenic signaling. These findings have important therapeutic implications for a variety of disorders involving abnormal angiogenesis and vascular leakage.
Figures



References
-
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. - PubMed
-
- Carmeliet P. Angiogenesis in health and disease. Nat. med. 2003;9:653–660. - PubMed
-
- Chang SR, Young BD, Li SJ, Qi XX, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321–334. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
