

Impacts of Usher syndrome type IB mutations on human myosin VIIa motor function
- PMID: 18700726
- PMCID: PMC2821024
- DOI: 10.1021/bi8007142
Impacts of Usher syndrome type IB mutations on human myosin VIIa motor function
Abstract
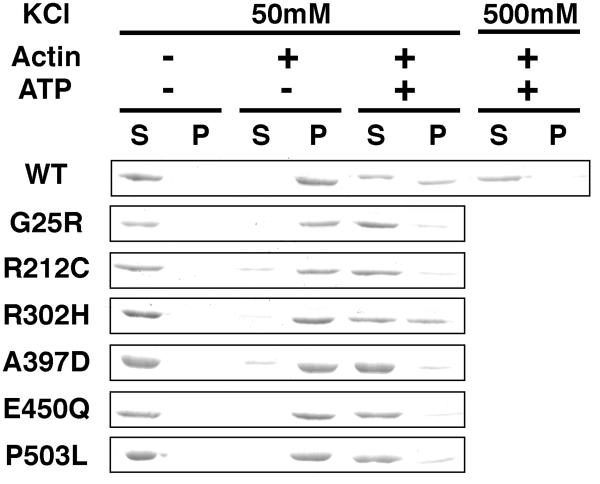
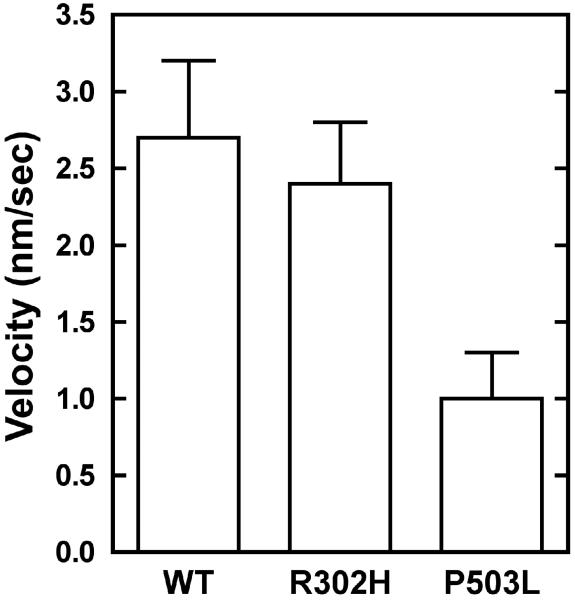
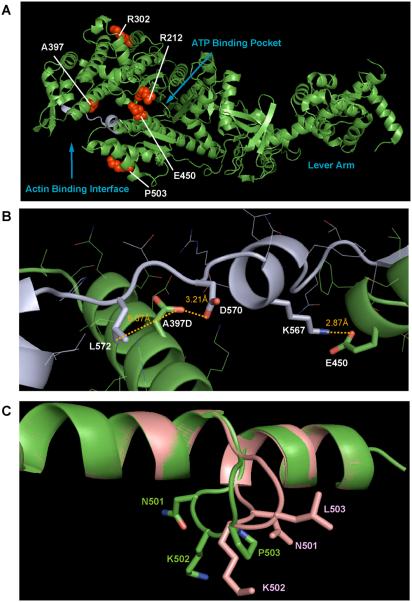
Usher syndrome (USH) is a human hereditary disorder characterized by profound congenital deafness, retinitis pigmentosa, and vestibular dysfunction. Myosin VIIa has been identified as the responsible gene for USH type 1B, and a number of missense mutations have been identified in the affected families. However, the molecular basis of the dysfunction of USH gene, myosin VIIa, in the affected families is unknown to date. Here we clarified the effects of USH1B mutations on human myosin VIIa motor function for the first time. The missense mutations of USH1B significantly inhibited the actin activation of ATPase activity of myosin VIIa. G25R, R212C, A397D, and E450Q mutations abolished the actin-activated ATPase activity completely. P503L mutation increased the basal ATPase activity for 2-3-fold but reduced the actin-activated ATPase activity to 50% of the wild type. While all of the mutations examined, except for R302H, reduced the affinity for actin and the ATP hydrolysis cycling rate, they did not largely decrease the rate of ADP release from actomyosin, suggesting that the mutations reduce the duty ratio of myosin VIIa. Taken together, the results suggest that the mutations responsible for USH1B cause the complete loss of the actin-activated ATPase activity or the reduction of duty ratio of myosin VIIa.
Figures
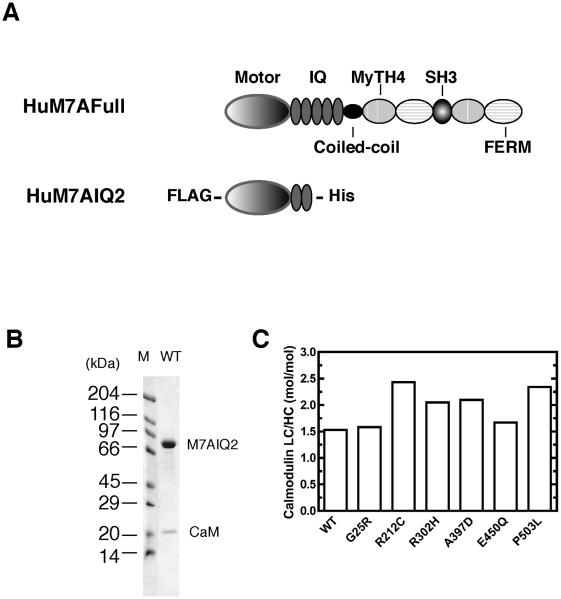
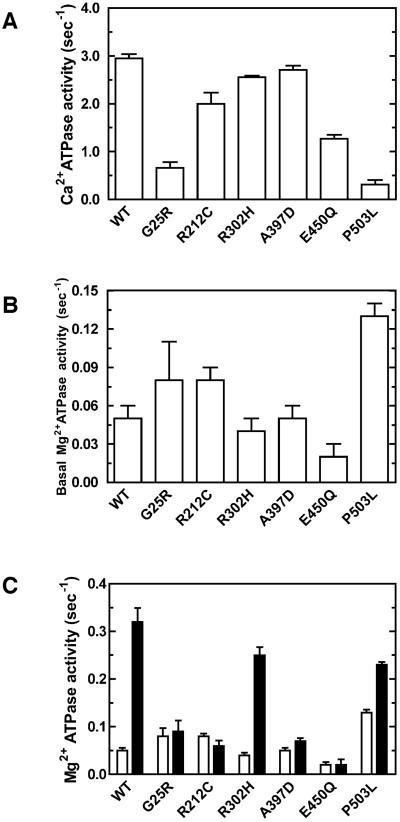
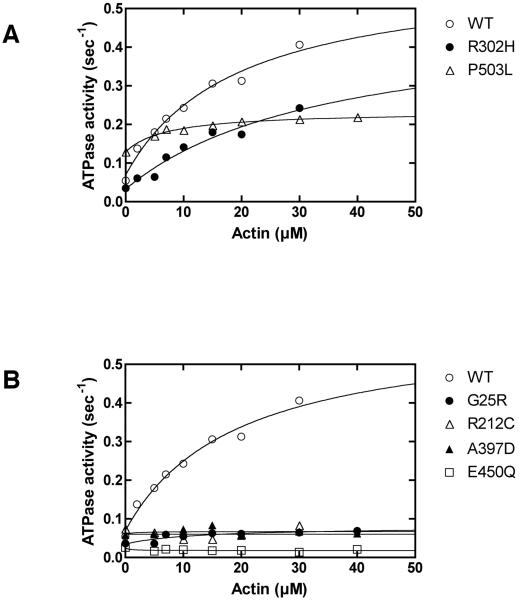
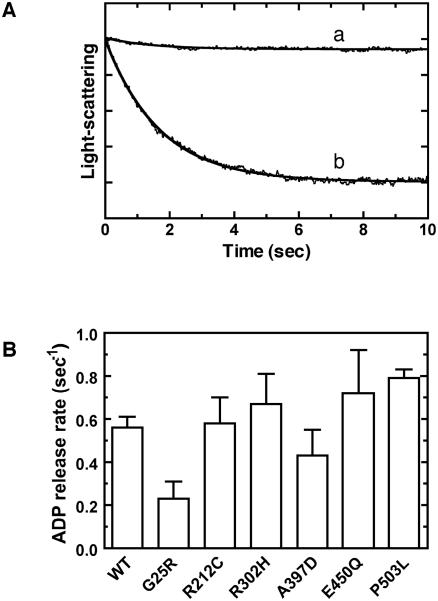
Similar articles
-
Drosophila myosin VIIA is a high duty ratio motor with a unique kinetic mechanism.J Biol Chem. 2006 Mar 17;281(11):7151-60. doi: 10.1074/jbc.M511592200. Epub 2006 Jan 16. J Biol Chem. 2006. PMID: 16415346
-
Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease.Exp Eye Res. 2006 Jul;83(1):97-119. doi: 10.1016/j.exer.2005.11.010. Epub 2006 Mar 20. Exp Eye Res. 2006. PMID: 16545802 Review.
-
The shaker-1 mouse myosin VIIa deafness mutation results in a severely reduced rate of the ATP hydrolysis step.J Biol Chem. 2018 Jan 19;293(3):819-829. doi: 10.1074/jbc.M117.810119. Epub 2017 Nov 22. J Biol Chem. 2018. PMID: 29167268 Free PMC article.
-
Vertebrate myosin VIIb is a high duty ratio motor adapted for generating and maintaining tension.J Biol Chem. 2005 Nov 25;280(47):39665-76. doi: 10.1074/jbc.M507667200. Epub 2005 Sep 26. J Biol Chem. 2005. PMID: 16186105
-
Genetics and pathological mechanisms of Usher syndrome.J Hum Genet. 2010 Jun;55(6):327-35. doi: 10.1038/jhg.2010.29. Epub 2010 Apr 9. J Hum Genet. 2010. PMID: 20379205 Free PMC article. Review.
Cited by
-
Human myosin VIIa is a very slow processive motor protein on various cellular actin structures.J Biol Chem. 2017 Jun 30;292(26):10950-10960. doi: 10.1074/jbc.M116.765966. Epub 2017 May 15. J Biol Chem. 2017. PMID: 28507101 Free PMC article.
-
Cargo binding activates myosin VIIA motor function in cells.Proc Natl Acad Sci U S A. 2011 Apr 26;108(17):7028-33. doi: 10.1073/pnas.1009188108. Epub 2011 Apr 11. Proc Natl Acad Sci U S A. 2011. PMID: 21482763 Free PMC article.
-
The tail binds to the head-neck domain, inhibiting ATPase activity of myosin VIIA.Proc Natl Acad Sci U S A. 2009 May 26;106(21):8483-8. doi: 10.1073/pnas.0812930106. Epub 2009 May 7. Proc Natl Acad Sci U S A. 2009. PMID: 19423668 Free PMC article.
-
The kinetic mechanism of mouse myosin VIIA.J Biol Chem. 2011 Mar 18;286(11):8819-28. doi: 10.1074/jbc.M110.163592. Epub 2011 Jan 6. J Biol Chem. 2011. PMID: 21212272 Free PMC article.
-
The Logistical Backbone of Photoreceptor Cell Function: Complementary Mechanisms of Dietary Vitamin A Receptors and Rhodopsin Transporters.Int J Mol Sci. 2024 Apr 12;25(8):4278. doi: 10.3390/ijms25084278. Int J Mol Sci. 2024. PMID: 38673863 Free PMC article. Review.
References
-
- Kaplan J, Gerber S, Bonneau D, Rozet JM, Delrieu O, Briard ML, Dollfus H, Ghazi I, Dufier JL, Frezal J, et al. A gene for Usher syndrome type I (USH1A) maps to chromosome 14q. Genomics. 1992;14:979–987. - PubMed
-
- Smith RJ, Lee EC, Kimberling WJ, Daiger SP, Pelias MZ, Keats BJ, Jay M, Bird A, Reardon W, Guest M, et al. Localization of two genes for Usher syndrome type I to chromosome 11. Genomics. 1992;14:995–1002. - PubMed
-
- Wayne S, Der Kaloustian VM, Schloss M, Polomeno R, Scott DA, Hejtmancik JF, Sheffield VC, Smith RJ. Localization of the Usher syndrome type ID gene (Ush1D) to chromosome 10. Hum Mol Genet. 1996;5:1689–1692. - PubMed
-
- Kimberling WJ, Moller CG, Davenport S, Priluck IA, Beighton PH, Greenberg J, Reardon W, Weston MD, Kenyon JB, Grunkemeyer JA, et al. Linkage of Usher syndrome type I gene (USH1B) to the long arm of chromosome 11. Genomics. 1992;14:988–994. - PubMed
-
- Chaib H, Kaplan J, Gerber S, Vincent C, Ayadi H, Slim R, Munnich A, Weissenbach J, Petit C. A newly identified locus for Usher syndrome type I, USH1E, maps to chromosome 21q21. Hum Mol Genet. 1997;6:27–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
