

The nuclear orphan receptor NR2F6 suppresses lymphocyte activation and T helper 17-dependent autoimmunity
- PMID: 18701084
- PMCID: PMC4941926
- DOI: 10.1016/j.immuni.2008.06.008
The nuclear orphan receptor NR2F6 suppresses lymphocyte activation and T helper 17-dependent autoimmunity
Abstract
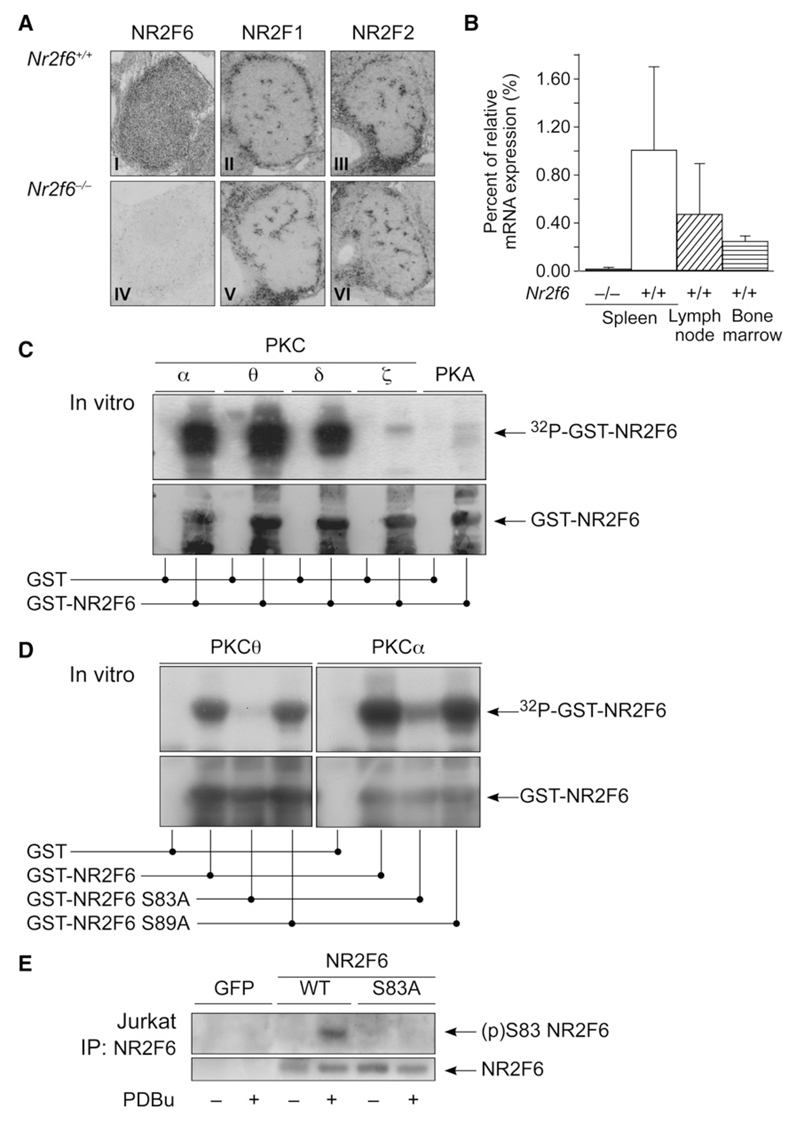
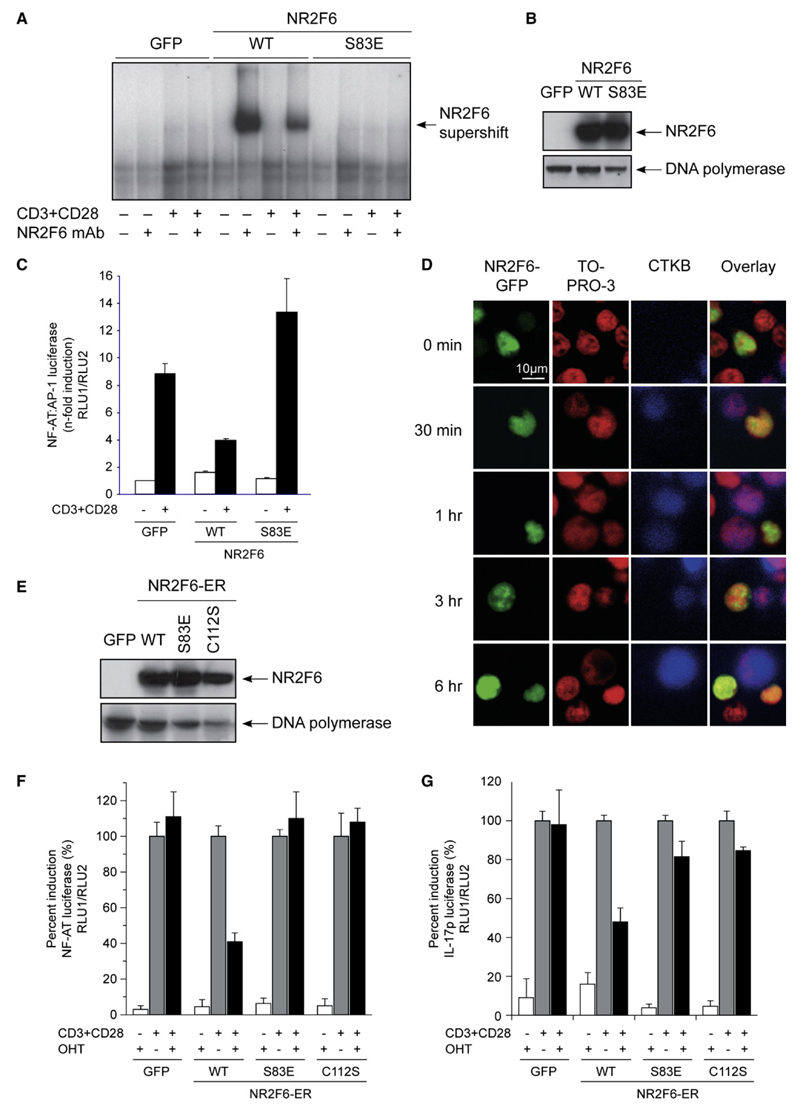
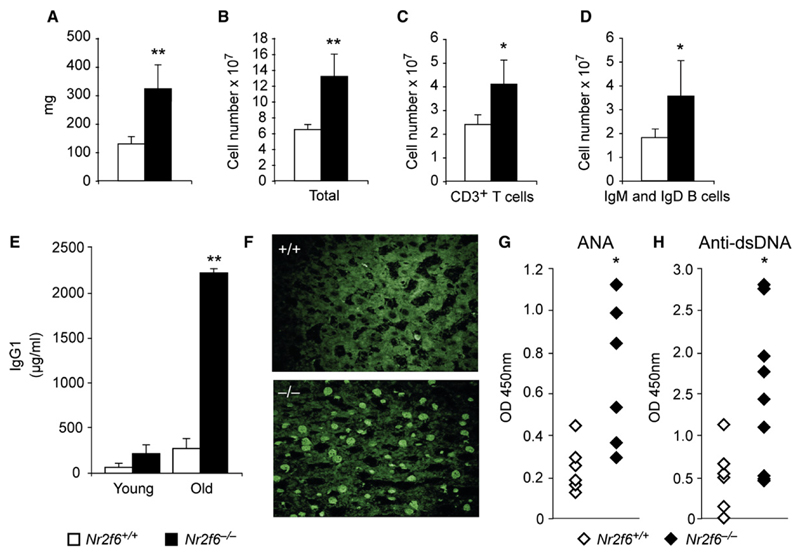
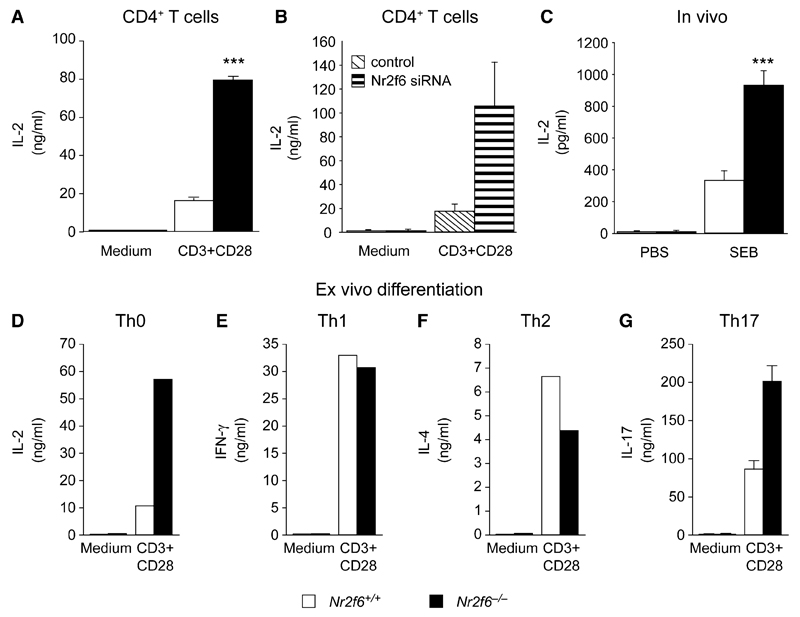
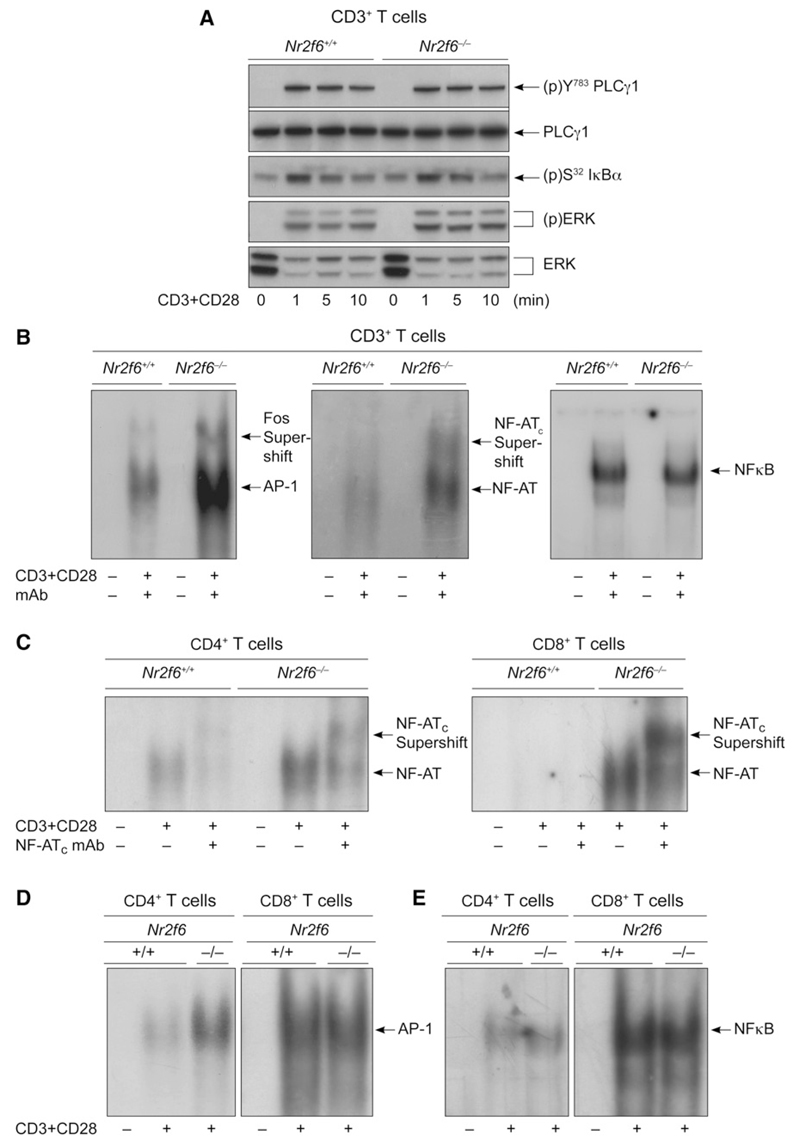
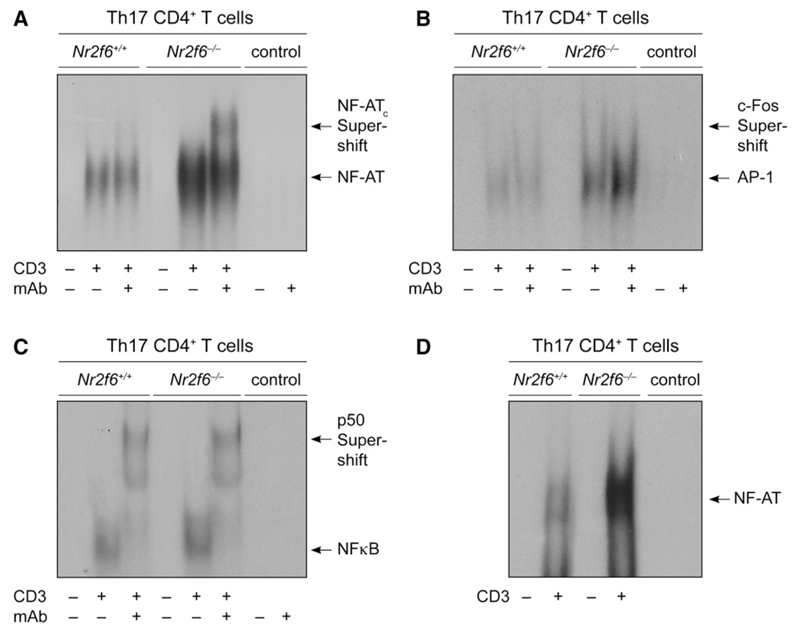
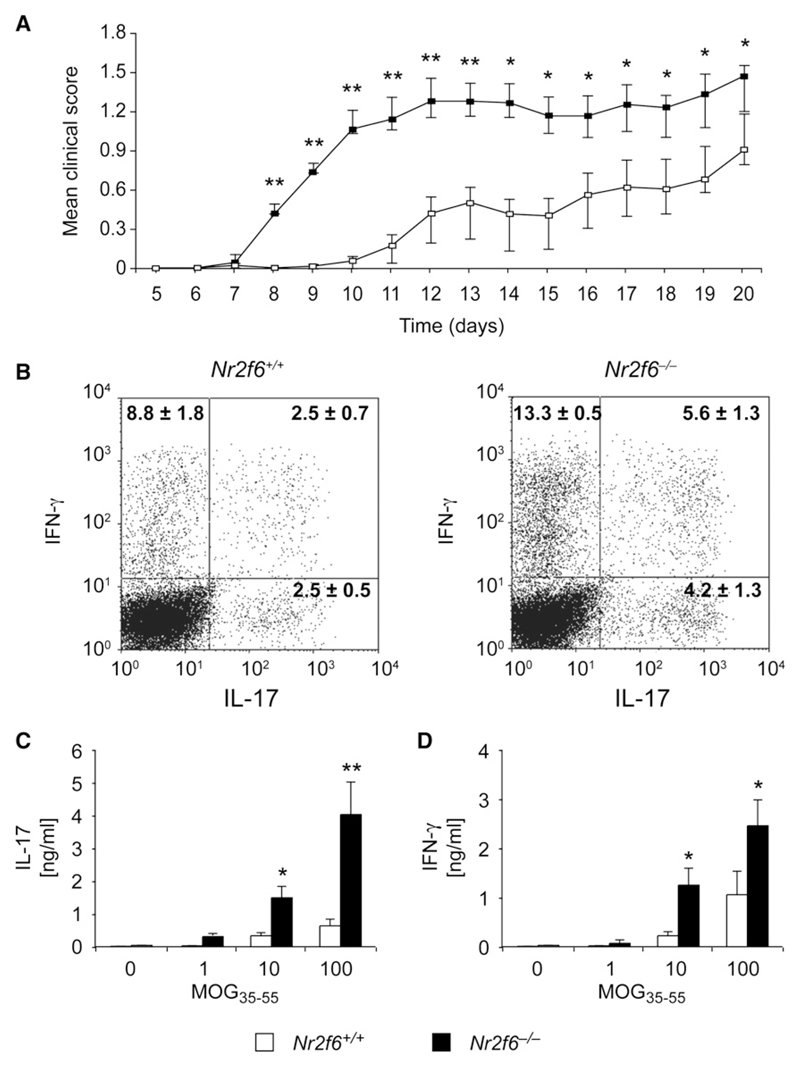
The protein kinase C (PKC) family of serine-threonine kinases plays a central role in T lymphocyte activation. Here, we identify NR2F6, a nuclear zinc-finger orphan receptor, as a critical PKC substrate and essential regulator of CD4(+) T cell activation responses. NR2F6 potently antagonized the ability of T helper 0 (Th0) and Th17 CD4(+) T cells to induce expression of key cytokine genes such as interleukin-2 (IL-2) and IL-17. Mechanistically, NR2F6 directly interfered with the DNA binding of nuclear factor of activated T cells (NF-AT):activator protein 1 (AP-1) but not nuclear factor kappaB (NF-kappa B) and, subsequently, transcriptional activity of the NF-AT-dependent IL-17A cytokine promoter. Consistent with our model, Nr2f6-deficient mice had hyperreactive lymphocytes, developed a late-onset immunopathology, and were hypersusceptible to Th17-dependent experimental autoimmune encephalomyelitis. Our study establishes NR2F6 as a transcriptional repressor of IL-17 expression in Th17-differentiated CD4(+) T cells in vitro and in vivo.
Figures
Comment in
-
Orphans against autoimmunity.Immunity. 2008 Aug 15;29(2):167-8. doi: 10.1016/j.immuni.2008.07.008. Immunity. 2008. PMID: 18701076
References
-
- Altman A, Kaminski S, Busuttil V, Droin N, Hu J, Hipskind RA, Villalba M. Positive feedback regulation of PLCγ1/Ca2+ signaling by PKCθ in restimulated T cells via a Tec kinase-dependent pathway. Eur J Immunol. 2004;34:2001–2011. - PubMed
-
- Anderson K, Fitzgerald M, Dupont M, Wang T, Paz N, Healy A, Xu Y, Ocain T, Schopf L, Jaffee B, et al. Mice deficient in PKCθ demonstrate impaired in vivo T cell activation and protection from T cell-mediated inflammatory diseases. Autoimmunity. 2006;39:469–478. - PubMed
-
- Baier G. The PKC gene module: Molecular biosystematics to resolve its T cell functions. Immunol Rev. 2003;192:64–79. - PubMed
-
- Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. - PubMed
-
- Chaudhary D, Kasaian M. PKCθ: A potential therapeutic target for T-cell-mediated diseases. Curr Opin Investig Drugs. 2006;7:432–437. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
