

Spectral dictionaries: Integrating de novo peptide sequencing with database search of tandem mass spectra
- PMID: 18703573
- PMCID: PMC2621003
- DOI: 10.1074/mcp.M800103-MCP200
Spectral dictionaries: Integrating de novo peptide sequencing with database search of tandem mass spectra
Abstract
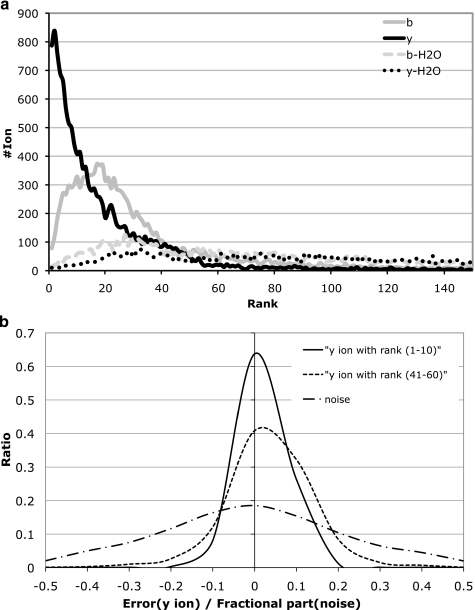
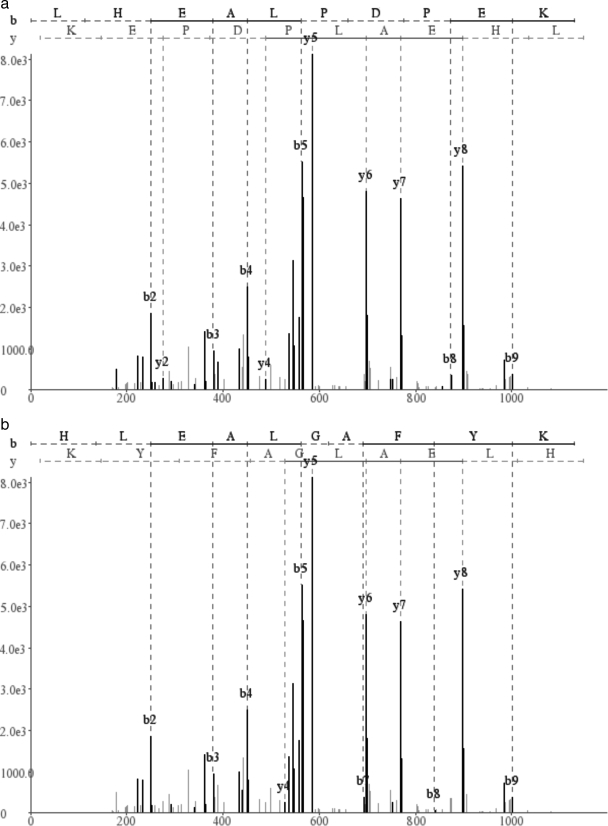
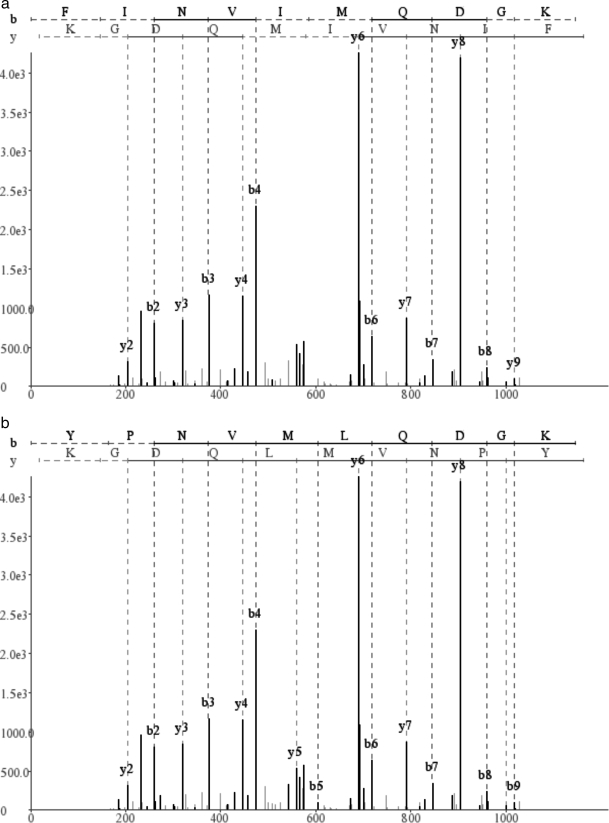
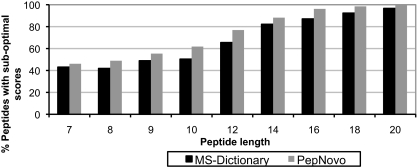
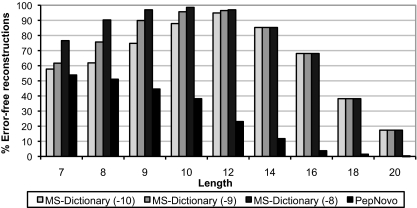
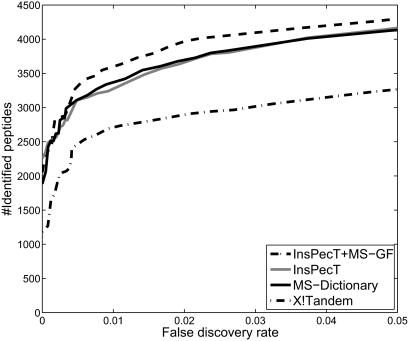
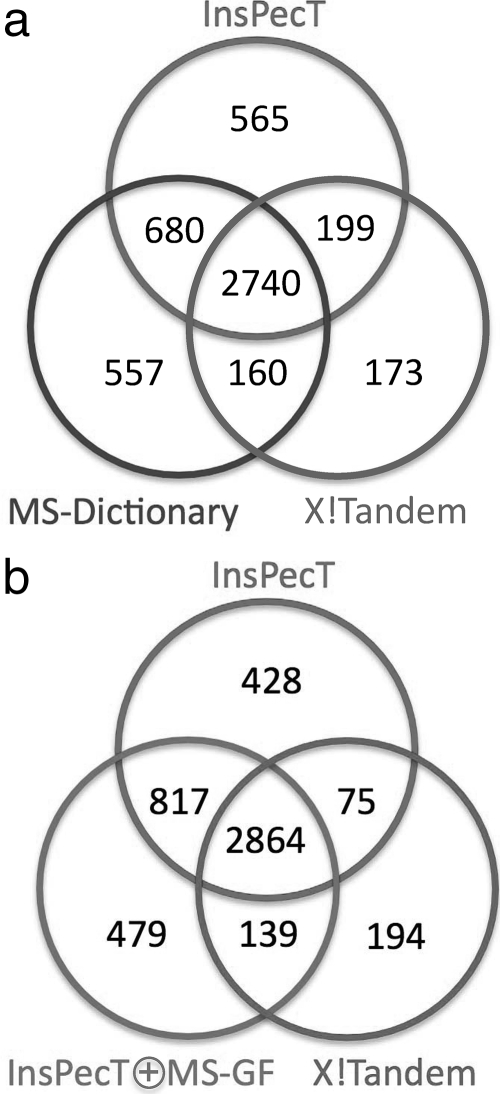
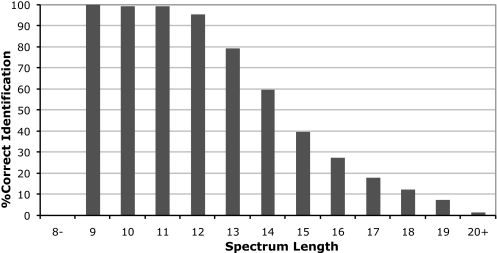
Database search tools identify peptides by matching tandem mass spectra against a protein database. We study an alternative approach when all plausible de novo interpretations of a spectrum (spectral dictionary) are generated and then quickly matched against the database. We present a new MS-Dictionary algorithm for efficiently generating spectral dictionaries and demonstrate that MS-Dictionary can identify spectra that are missed in the database search. We argue that MS-Dictionary enables proteogenomics searches in six-frame translation of genomic sequences that may be prohibitively time-consuming for existing database search approaches. We show that such searches allow one to correct sequencing errors and find programmed frameshifts.
Figures
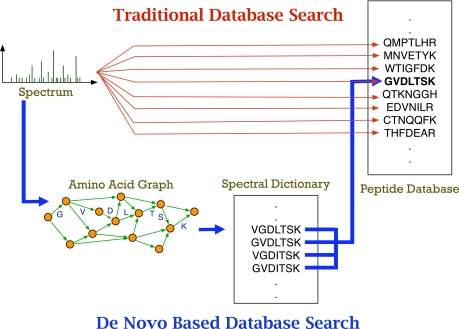
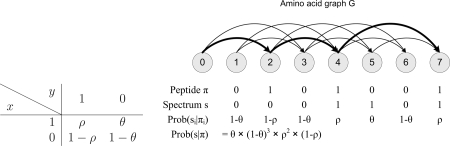
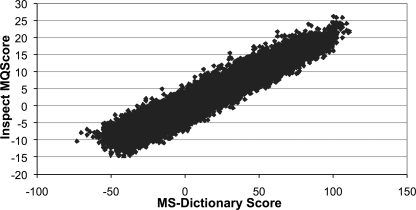
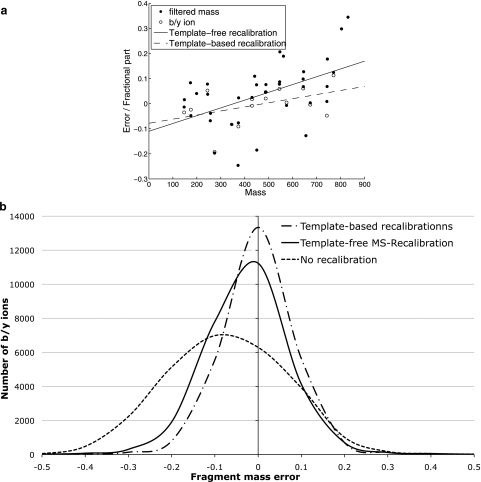
References
-
- Mann, M., and Wilm, M. ( 1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 - PubMed
-
- Tanner, S., Shu, H., Frank, A., Wang, L., Zandi, E., Mumby, M., Pevzner, P., and Bafna, V. ( 2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 - PubMed
-
- Shilov, I., Seymour, S., Patel, A., Loboda, A., Tang, W., Keating, S., Hunter, C., Nuwaysir, L., and Schaeffer, D. ( 2007) The Paragon Algorithm: a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 - PubMed
-
- Frank, A., Tanner, S., Bafna, V., and Pevzner, P. ( 2005) Peptide sequence tags for fast database search in mass-spectrometry. J. Proteome Res. 4, 1287–1295 - PubMed
-
- Liu, C., Yan, B., Song, Y., Xu, Y., and Cai, L. ( 2006) Peptide sequence tag-based blind identification of post-translational modifications with point process model. Bioinformatics 22, e307–e313 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
