

Role of small GTPases and alphavbeta5 integrin in Pseudomonas aeruginosa-induced increase in lung endothelial permeability
- PMID: 18703797
- PMCID: PMC2606944
- DOI: 10.1165/rcmb.2007-0454OC
Role of small GTPases and alphavbeta5 integrin in Pseudomonas aeruginosa-induced increase in lung endothelial permeability
Abstract
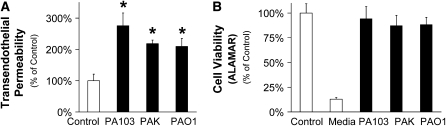
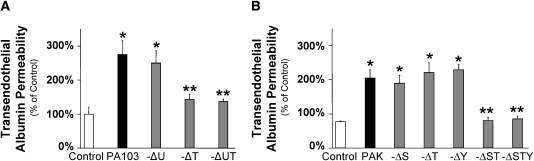
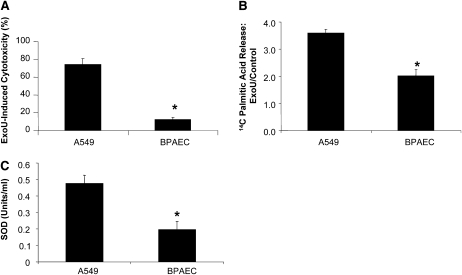
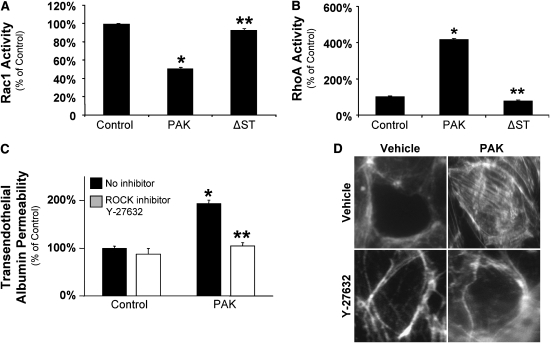
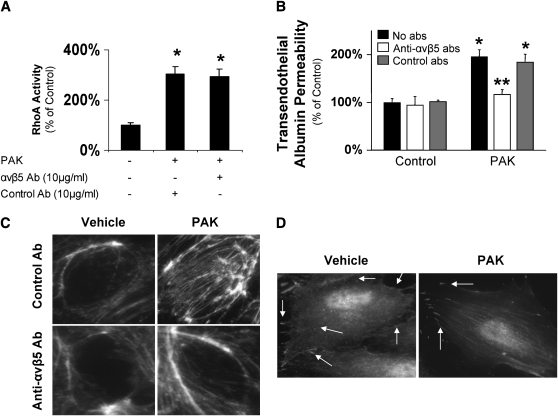
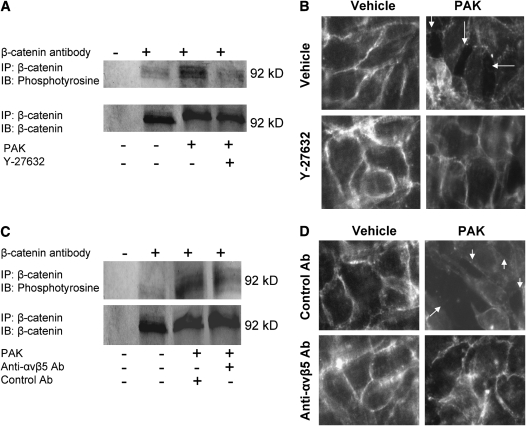
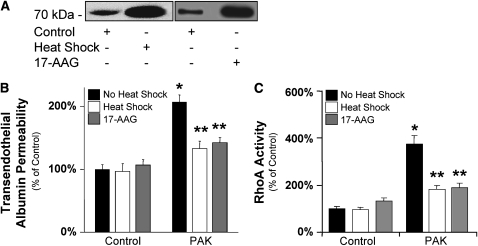
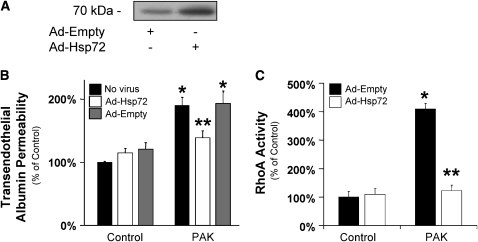
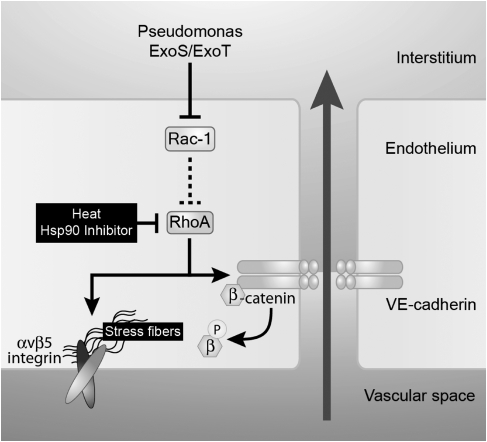
Pseudomonas aeruginosa is an opportunistic pathogen that can cause severe pneumonia associated with airspace flooding with protein-rich edema in critically ill patients. The type III secretion system is a major virulence factor and contributes to dissemination of P. aeruginosa. However, it is still unknown which particular bacterial toxin and which cellular pathways are responsible for the increase in lung endothelial permeability induced by P. aeruginosa. Thus, the first objective of this study was to determine the mechanisms by which this species causes an increase in lung endothelial permeability. The results showed that ExoS and ExoT, two of the four known P. aeruginosa type III cytotoxins, were primarily responsible for bacterium-induced increases in protein permeability across the lung endothelium via an inhibition of Rac1 and an activation of the RhoA signaling pathway. In addition, inhibition of the alphavbeta5 integrin, a central regulator of lung vascular permeability, prevented these P. aeruginosa-mediated increases in albumin flux due to endothelial permeability. Finally, prior activation of the stress protein response or adenoviral gene transfer of the inducible heat shock protein Hsp72 also inhibited the damaging effects of P. aeruginosa on the barrier function of lung endothelium. Taken together, these results demonstrate the critical role of the RhoA/alphavbeta5 integrin pathway in mediating P. aeruginosa-induced lung vascular permeability. In addition, activation of the stress protein response with pharmacologic inhibitors of Hsp90 may protect lungs against P. aeruginosa-induced permeability changes.
Figures
Similar articles
-
Cytoprotective-selective activated protein C attenuates Pseudomonas aeruginosa-induced lung injury in mice.Am J Respir Cell Mol Biol. 2011 Sep;45(3):632-41. doi: 10.1165/rcmb.2010-0397OC. Epub 2011 Jan 21. Am J Respir Cell Mol Biol. 2011. PMID: 21257925 Free PMC article.
-
In vivo rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS.Infect Immun. 2002 Jan;70(1):360-7. doi: 10.1128/IAI.70.1.360-367.2002. Infect Immun. 2002. PMID: 11748202 Free PMC article.
-
Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42.Infect Immun. 2002 Apr;70(4):2198-205. doi: 10.1128/IAI.70.4.2198-2205.2002. Infect Immun. 2002. PMID: 11895987 Free PMC article.
-
Pseudomonas aeruginosa ExoS and ExoT.Rev Physiol Biochem Pharmacol. 2004;152:79-92. doi: 10.1007/s10254-004-0031-7. Epub 2004 Aug 24. Rev Physiol Biochem Pharmacol. 2004. PMID: 15375697 Review.
-
Role of Pseudomonas aeruginosa type III effectors in disease.Curr Opin Microbiol. 2009 Feb;12(1):61-6. doi: 10.1016/j.mib.2008.12.007. Epub 2009 Jan 23. Curr Opin Microbiol. 2009. PMID: 19168385 Review.
Cited by
-
Molecular Mechanisms Involved in Pseudomonas aeruginosa Bacteremia.Adv Exp Med Biol. 2022;1386:325-345. doi: 10.1007/978-3-031-08491-1_12. Adv Exp Med Biol. 2022. PMID: 36258078 Review.
-
The Role of Pseudomonas aeruginosa Virulence Factors in Cytoskeletal Dysregulation and Lung Barrier Dysfunction.Toxins (Basel). 2021 Nov 2;13(11):776. doi: 10.3390/toxins13110776. Toxins (Basel). 2021. PMID: 34822560 Free PMC article. Review.
-
Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa.Am J Physiol Lung Cell Mol Physiol. 2014 Apr 1;306(7):L591-603. doi: 10.1152/ajplung.00335.2013. Epub 2014 Jan 24. Am J Physiol Lung Cell Mol Physiol. 2014. PMID: 24464809 Free PMC article. Review.
-
In the absence of effector proteins, the Pseudomonas aeruginosa type three secretion system needle tip complex contributes to lung injury and systemic inflammatory responses.PLoS One. 2013 Nov 27;8(11):e81792. doi: 10.1371/journal.pone.0081792. eCollection 2013. PLoS One. 2013. PMID: 24312357 Free PMC article.
-
The extreme C terminus of the Pseudomonas aeruginosa effector ExoY is crucial for binding to its eukaryotic activator, F-actin.J Biol Chem. 2018 Dec 21;293(51):19785-19796. doi: 10.1074/jbc.RA118.003784. Epub 2018 Oct 30. J Biol Chem. 2018. PMID: 30377256 Free PMC article.
References
-
- Rello J, Rue M, Jubert P, Muses G, Sonora R, Valles J, Niederman MS. Survival in patients with nosocomial pneumonia: impact of the severity of illness and the etiologic agent. Crit Care Med 1997;25:1862–1867. - PubMed
-
- Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, Wu C, Mende-Mueller L, Frank DW. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol 1997;25:547–557. - PubMed
-
- Kipnis E, Sawa T, Wiener-Kronish J. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med Mal Infect 2006;36:78–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
