

Genetic variation in an individual human exome
- PMID: 18704161
- PMCID: PMC2493042
- DOI: 10.1371/journal.pgen.1000160
Genetic variation in an individual human exome
Abstract
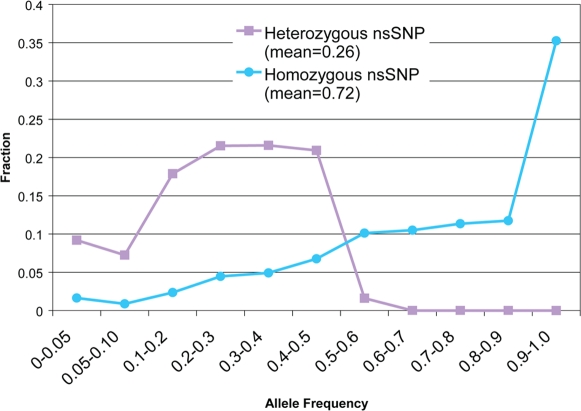
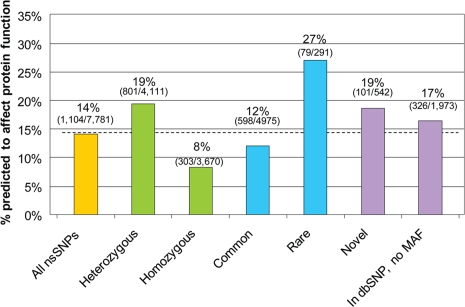
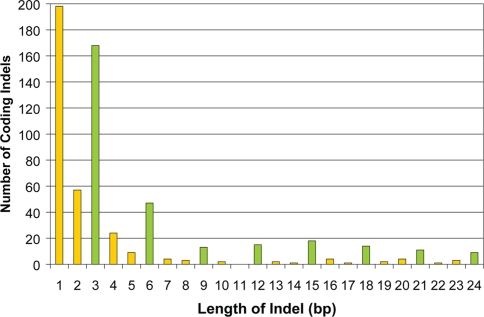
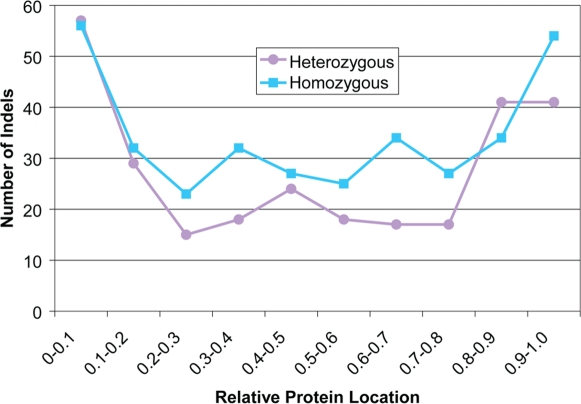
There is much interest in characterizing the variation in a human individual, because this may elucidate what contributes significantly to a person's phenotype, thereby enabling personalized genomics. We focus here on the variants in a person's 'exome,' which is the set of exons in a genome, because the exome is believed to harbor much of the functional variation. We provide an analysis of the approximately 12,500 variants that affect the protein coding portion of an individual's genome. We identified approximately 10,400 nonsynonymous single nucleotide polymorphisms (nsSNPs) in this individual, of which approximately 15-20% are rare in the human population. We predict approximately 1,500 nsSNPs affect protein function and these tend be heterozygous, rare, or novel. Of the approximately 700 coding indels, approximately half tend to have lengths that are a multiple of three, which causes insertions/deletions of amino acids in the corresponding protein, rather than introducing frameshifts. Coding indels also occur frequently at the termini of genes, so even if an indel causes a frameshift, an alternative start or stop site in the gene can still be used to make a functional protein. In summary, we reduced the set of approximately 12,500 nonsilent coding variants by approximately 8-fold to a set of variants that are most likely to have major effects on their proteins' functions. This is our first glimpse of an individual's exome and a snapshot of the current state of personalized genomics. The majority of coding variants in this individual are common and appear to be functionally neutral. Our results also indicate that some variants can be used to improve the current NCBI human reference genome. As more genomes are sequenced, many rare variants and non-SNP variants will be discovered. We present an approach to analyze the coding variation in humans by proposing multiple bioinformatic methods to hone in on possible functional variation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
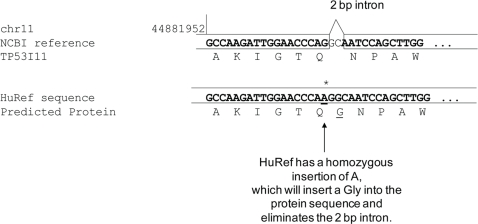
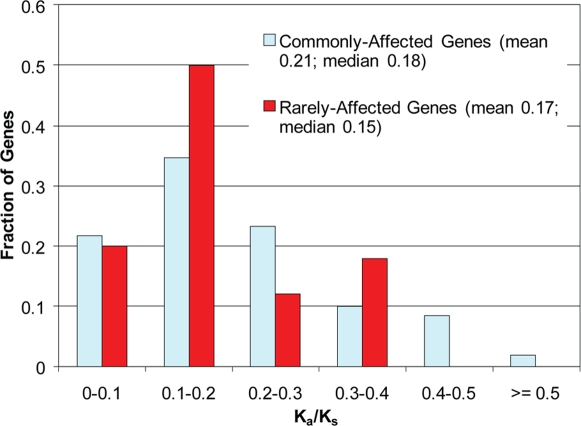
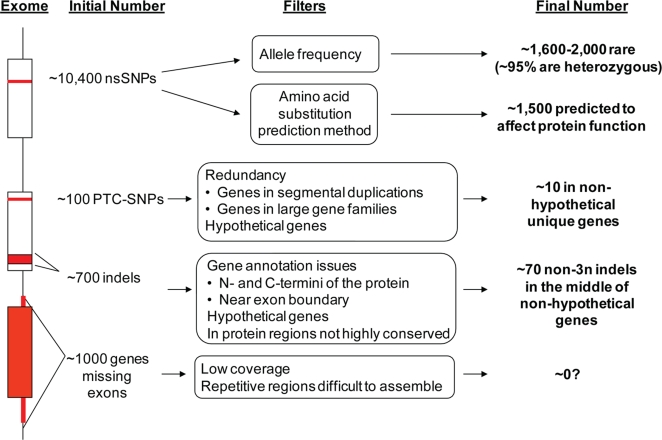
References
-
- Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003;33(Suppl):228–237. - PubMed
-
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. - PubMed
-
- Chakravarti A. Population genetics–making sense out of sequence. Nat Genet. 1999;21:56–60. - PubMed
-
- Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. - PubMed
-
- Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, et al. Haplotype variation and linkage disequilibrium in 313 human genes. Science. 2001;293:489–493. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
