

Detection of infectious prions in urine
- PMID: 18706416
- PMCID: PMC2593137
- DOI: 10.1016/j.febslet.2008.08.003
Detection of infectious prions in urine
Abstract
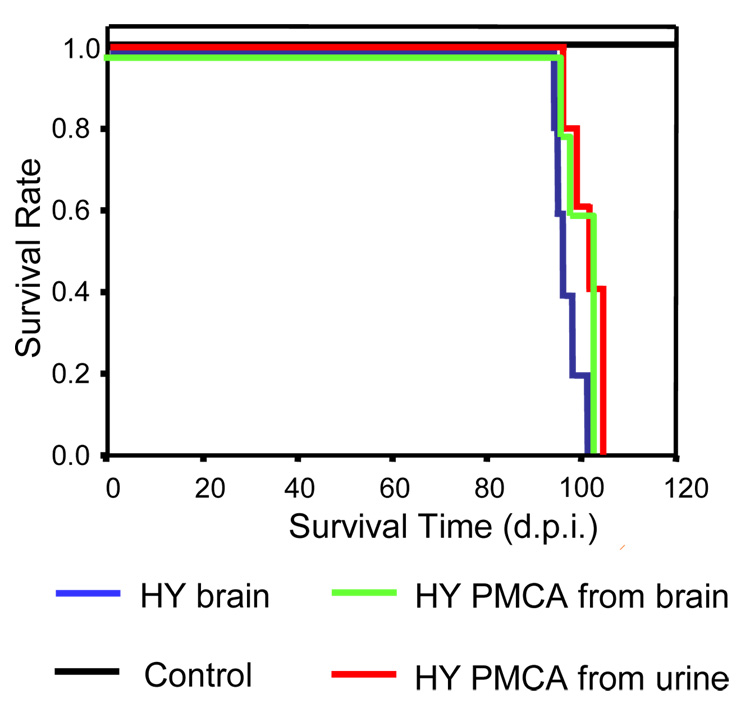
Prions are the infectious agents responsible for prion diseases, which appear to be composed exclusively by the misfolded prion protein (PrP(Sc)). The mechanism of prion transmission is unknown. In this study, we attempted to detect prions in urine of experimentally infected animals. PrP(Sc) was detected in approximately 80% of the animals studied, whereas no false positives were observed among the control animals. Semi-quantitative calculations suggest that PrP(Sc) concentration in urine is around 10-fold lower than in blood. Interestingly, PrP(Sc) present in urine maintains its infectious properties. Our data indicate that low quantities of infectious prions are excreted in the urine. These findings suggest that urine is a possible source of prion transmission.
Figures



References
-
- Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. - PubMed
-
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 2001;24:519–550. - PubMed
-
- Wadsworth JD, Collinge J. Update on human prion disease. Biochim. Biophys. Acta. 2007;1772:598–609. - PubMed
-
- Llewellyn CA, Hewitt P, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
