

Molecular basis for the structural instability of human DJ-1 induced by the L166P mutation associated with Parkinson's disease
- PMID: 18707128
- PMCID: PMC2646841
- DOI: 10.1021/bi800677k
Molecular basis for the structural instability of human DJ-1 induced by the L166P mutation associated with Parkinson's disease
Abstract
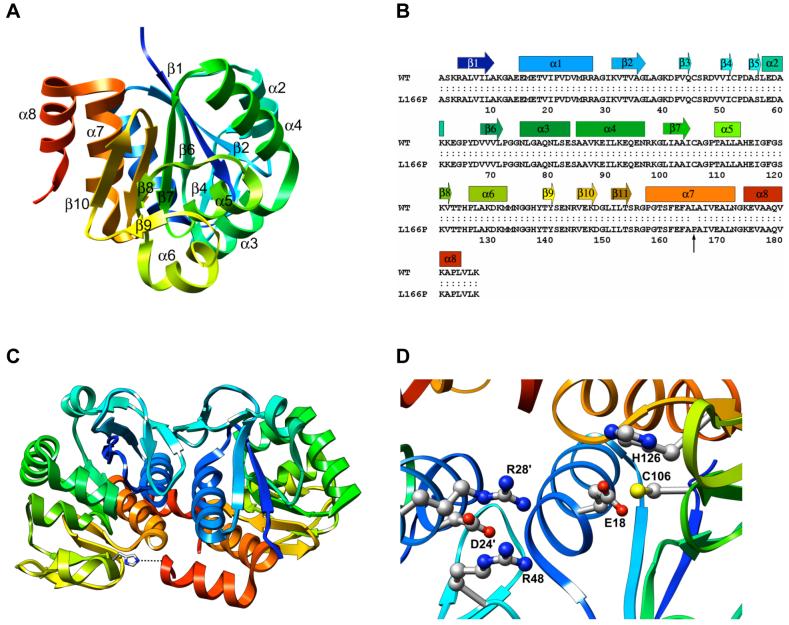
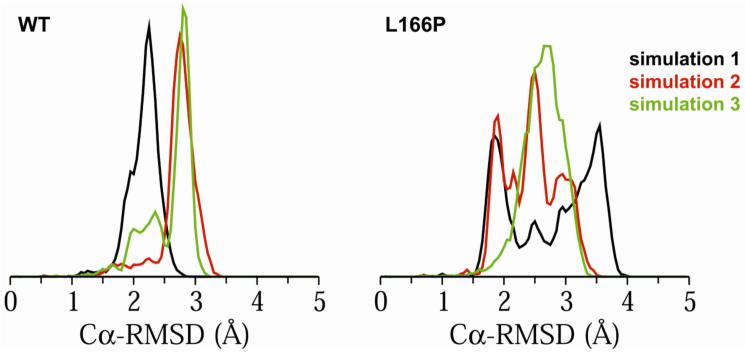
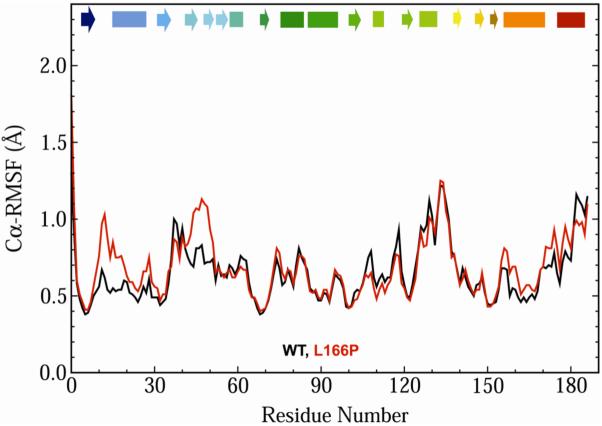
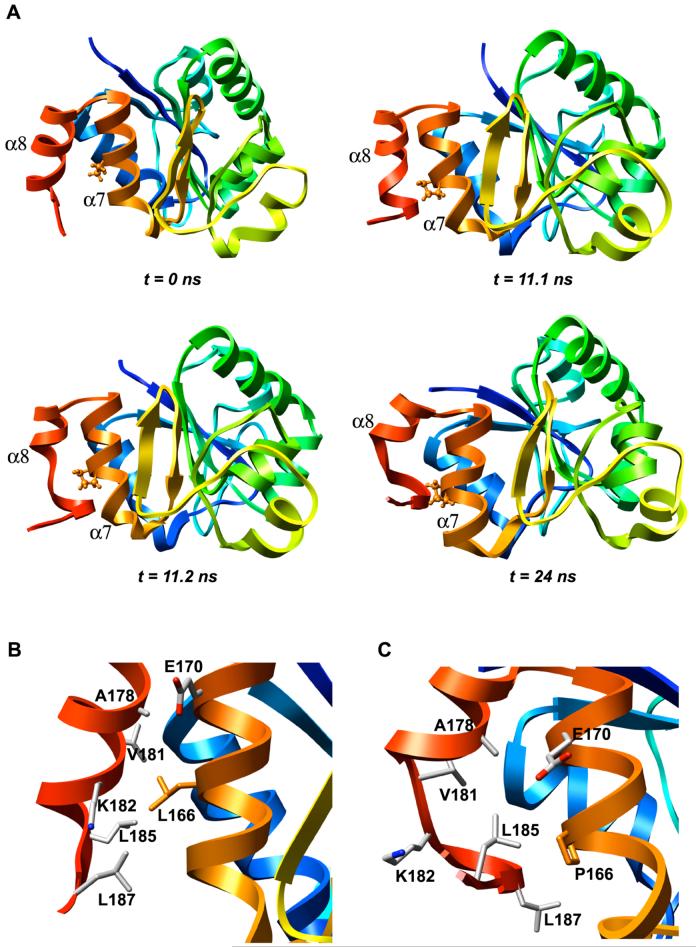
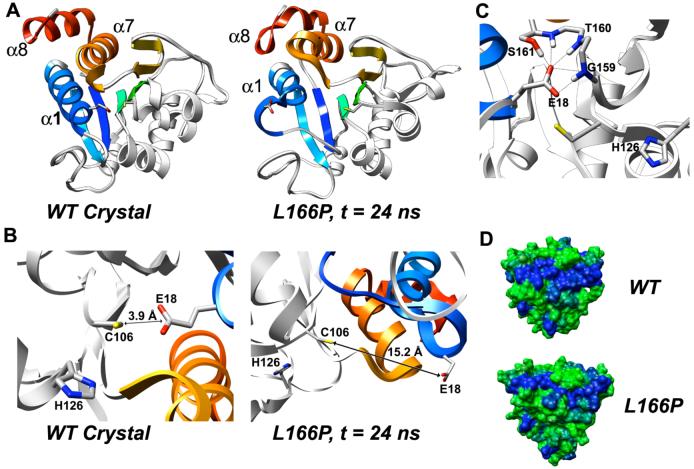
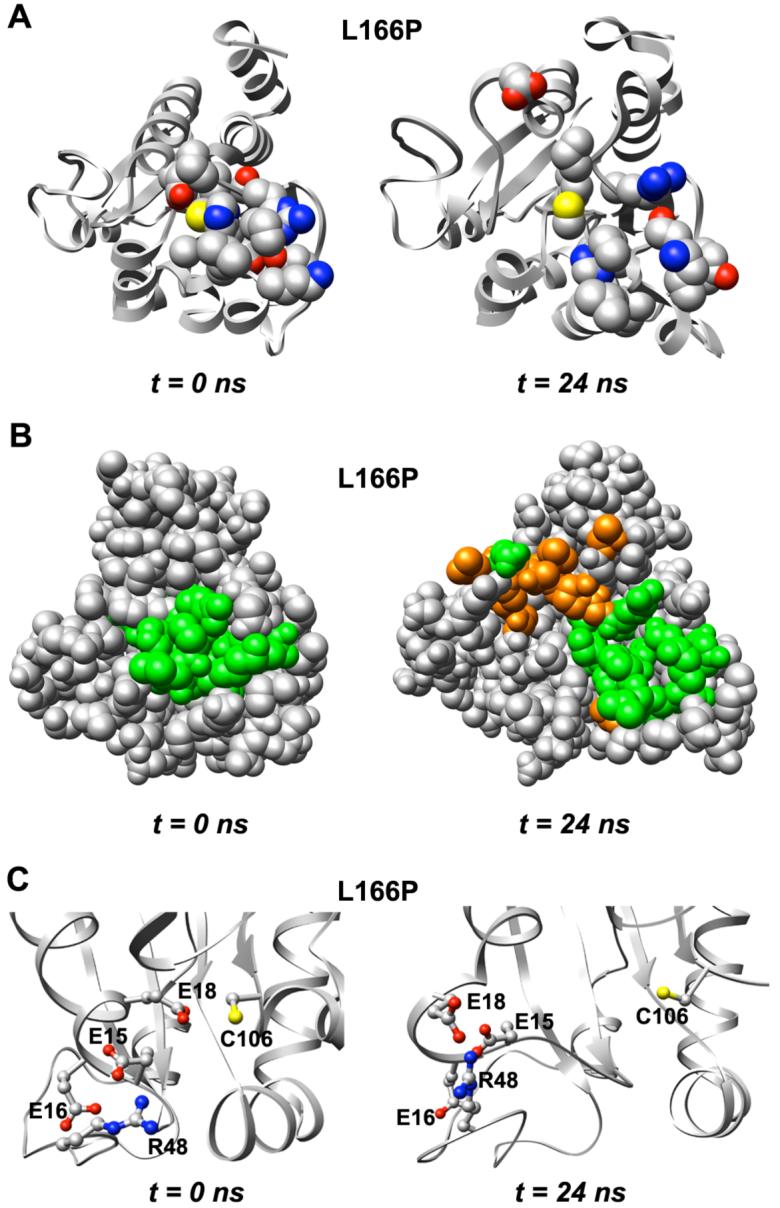
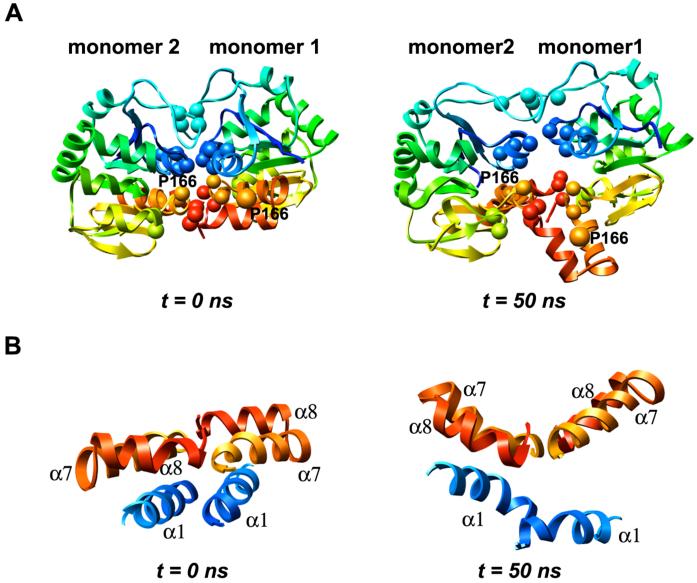
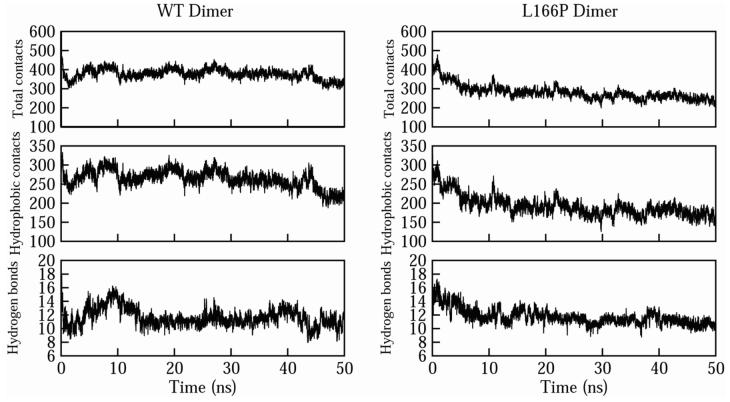
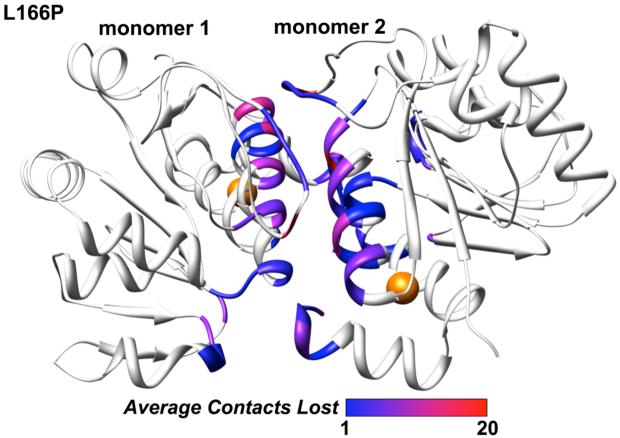
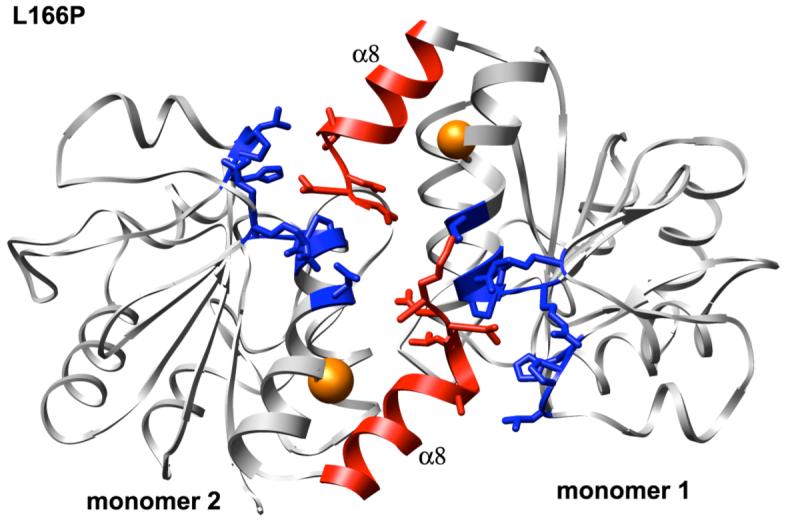
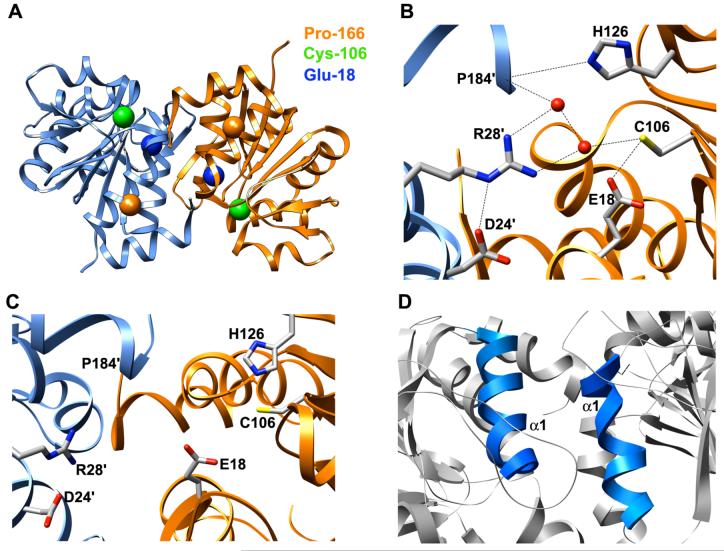
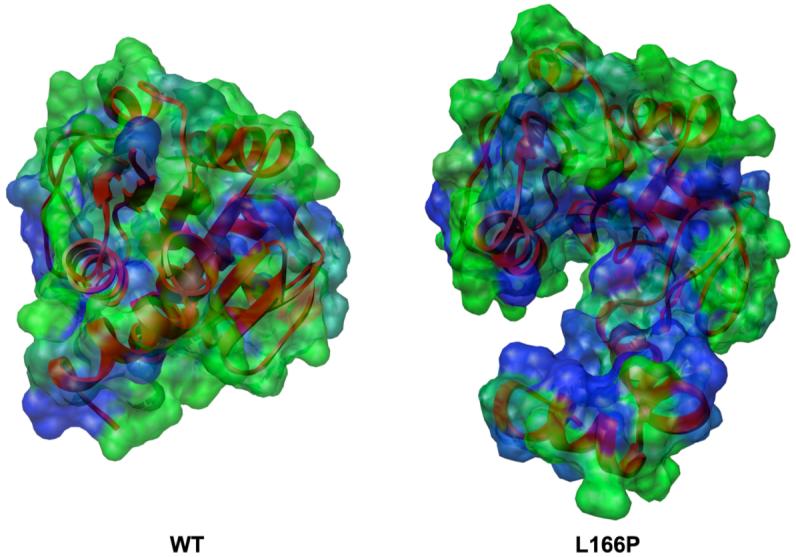
DJ-1 is a dimeric protein of unknown function in vivo. A mutation in the human DJ-1 gene causing substitution of proline for leucine at residue 166 (L166P) has been linked to early onset Parkinson's disease. Lack of structural stability has precluded experimental determination of atomic-resolution structures of the L166P DJ-1 polymorph. We have performed multiple molecular dynamics (MD) simulations ( approximately 1/3 mus) of the wild-type and L166P DJ-1 polymorph at physiological temperature to predict specific structural effects of the L166P substitution. L166P disrupted helices alpha1, alpha5, alpha6 and alpha8 with alpha8 undergoing particularly severe disruption. Secondary structural elements critical for protein stability and dimerization were significantly disrupted across the entire dimer interface, as were extended hydrophobic surfaces involved in dimer formation. Relative to wild-type DJ-1, L166P DJ-1 populated a broader ensemble of structures, many of which corresponded to distorted conformations. In a L166P dimer model the substitution significantly destabilized the dimer interface, interrupting >100 intermolecular contacts that are important for dimer formation. The L166P substitution also led to major perturbations in the region of a highly conserved cysteine residue (Cys-106) that participates in dimerization and that is critical for a proposed chaperone function of DJ-1. Cys-106 is located approximately 16 A from the substitution site, demonstrating that structural disruptions propagate throughout the whole protein. Furthermore, L166P DJ-1 showed a significant increase in hydrophobic surface area relative to wild-type protein, possibly explaining the tendency of the mutant protein to aggregate. These simulations provide details about specific structural disturbances throughout L166P DJ-1 that previous studies have not revealed.
Figures

References
-
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat. Rev. Genet. 2006;7:306–318. - PubMed
-
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. - PubMed
-
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. - PubMed
-
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000;25:302–305. - PubMed
-
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–452. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
