

Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways
- PMID: 18712785
- PMCID: PMC4118693
- DOI: 10.1002/hep.22397
Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways
Abstract
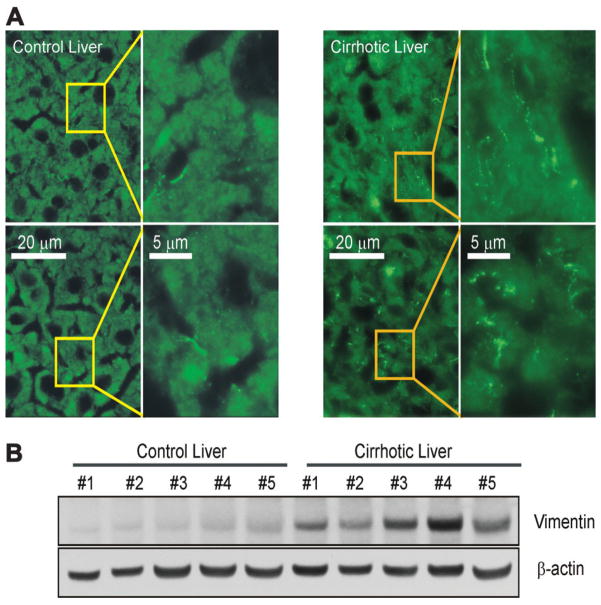
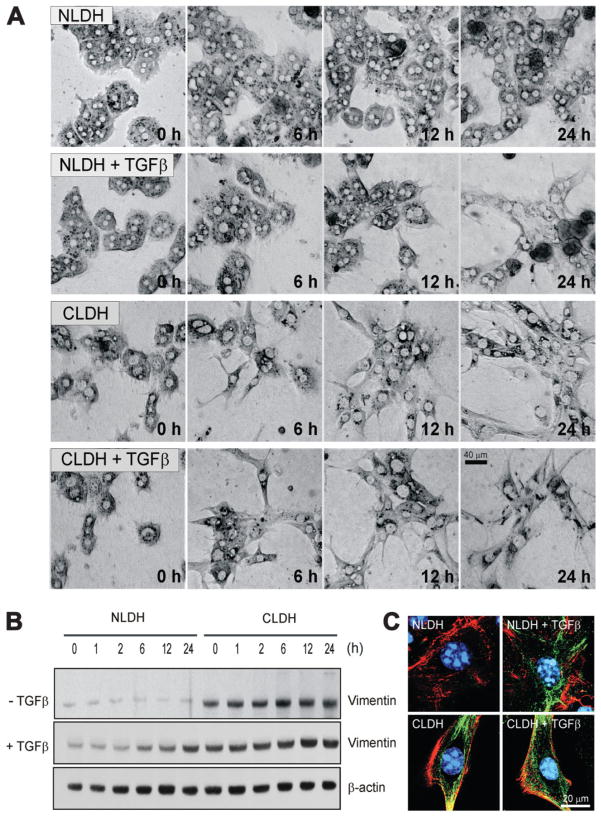
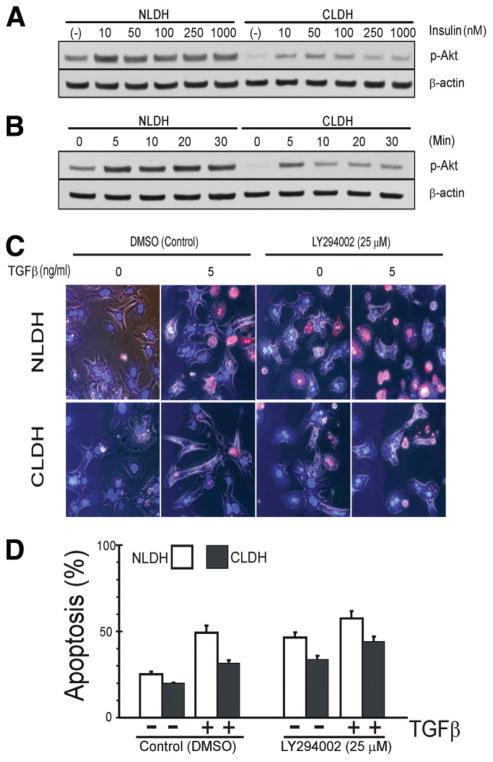
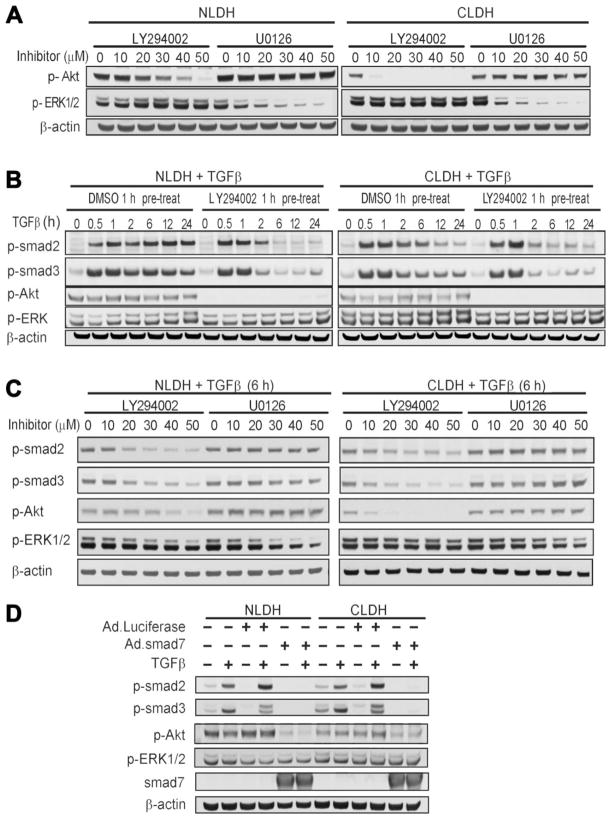
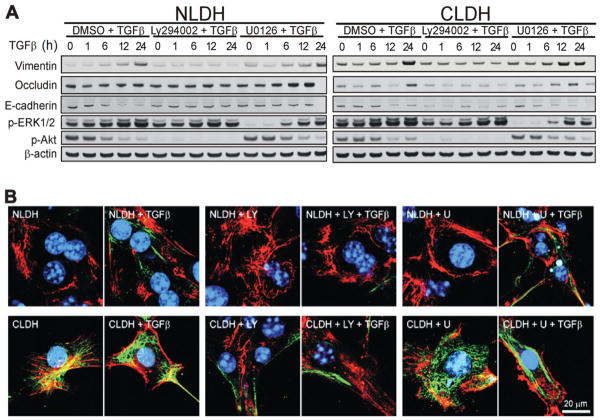
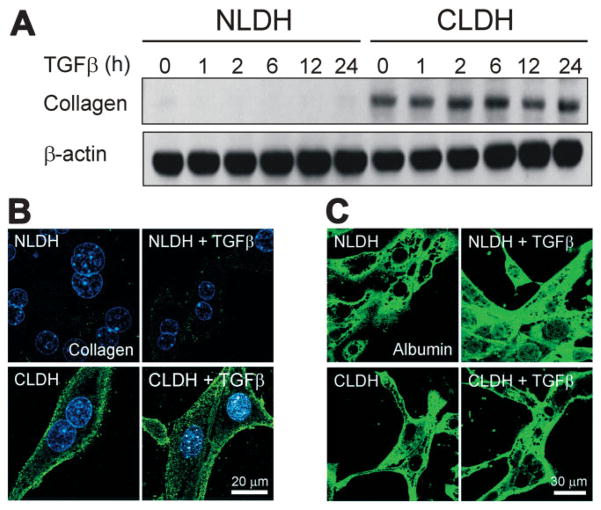
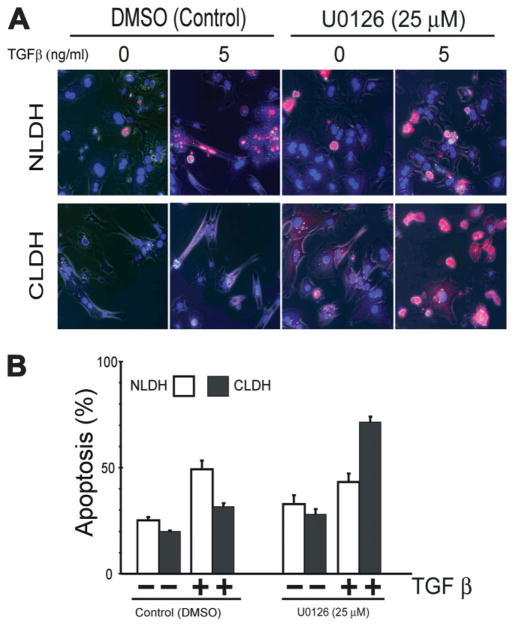
Hepatocytes that reside in a chronically-injured liver have altered growth responses compared to hepatocytes in normal liver. Transforming growth factor beta (TGFbeta) is upregulated in the cirrhotic liver, and cirrhotic hepatocytes, unlike normal hepatocytes exposed to this cytokine, exhibit decreased apoptosis. In fetal hepatocytes, TGFbeta also induces epithelial-mesenchymal transition (EMT) and signaling changes in cell survival pathways. Here, chronic murine liver injury was induced by twice-weekly carbon tetrachloride administration for 8 weeks. Normal liver-derived hepatocytes (NLDH) and cirrhotic liver-derived hepatocytes (CLDH) were examined for EMT and the small mothers against decapentaplegic homolog (Smad), phosphatidylinositol-3-kinase (PI3K/Akt), and mitogen activated protein kinase (MAPK) pathways were investigated. Immunofluorescence imaging of cirrhotic livers demonstrated increased vimentin expression, which was confirmed by immunoblot analysis. In vitro, CLDH exhibited increased vimentin and type 1 collagen expression within cellular extensions consistent with EMT. Treatment with TGFbeta augmented the EMT response in CLDH. In contrast, untreated NLDH did not display features of EMT but responded to TGFbeta with increased vimentin expression and EMT characteristics. In response to PI3K/Akt inhibition, CLDH had decreased basal and insulin-stimulated p-Akt expression and decreased apoptosis compared to NLDH. In both NLDH and CLDH, vimentin expression was dependent on PI3K/Akt activity. CLDH demonstrated increased basal p-extracellular signal-regulated kinase expression that was independent of Smad and PI3K/Akt signaling. Inhibition of the MAPK pathway produced a marked increase in CLDH apoptosis.
Conclusion: CLDH have increased vimentin and type 1 collagen expression and morphologic features consistent with EMT. In addition, compared to NLDH, the cellular signaling phenotype of CLDH changes from a MAPK-independent pathway to a MAPK-dependent cell survival pathway. These findings may have clinical implications for chemoprevention of hepatocellular carcinoma in the cirrhotic liver.
Conflict of interest statement
Potential conflict of interest: Nothing to report.
Figures
References
-
- Eghbali-Fatourechi G, Sieck GC, Prakash YS, Maercklein P, Gores GJ, Fitzpatrick LA. Type I procollagen production and cell proliferation is mediated by transforming growth factor-beta in a model of hepatic fibrosis. Endocrinology. 1996;137:1894–1903. - PubMed
-
- Schrum LW, Bird MA, Salcher O, Burchardt ER, Grisham JW, Brenner DA, et al. Autocrine expression of activated transforming growth factor-beta(1) induces apoptosis in normal rat liver. Am J Physiol Gastrointest Liver Physiol. 2001;280:G139–G148. - PubMed
-
- Black D, Bird MA, Samson CM, Lyman S, Lange PA, Schrum LW, et al. Primary cirrhotic hepatocytes resist TGFbeta-induced apoptosis through a ROS-dependent mechanism. J Hepatol. 2004;40:942–951. - PubMed
-
- Valdes F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernandez M, et al. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res. 2002;1:68–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical