

Dehydroepiandrosterone stimulates phosphorylation of FoxO1 in vascular endothelial cells via phosphatidylinositol 3-kinase- and protein kinase A-dependent signaling pathways to regulate ET-1 synthesis and secretion
- PMID: 18718910
- PMCID: PMC2570854
- DOI: 10.1074/jbc.M802906200
Dehydroepiandrosterone stimulates phosphorylation of FoxO1 in vascular endothelial cells via phosphatidylinositol 3-kinase- and protein kinase A-dependent signaling pathways to regulate ET-1 synthesis and secretion
Abstract
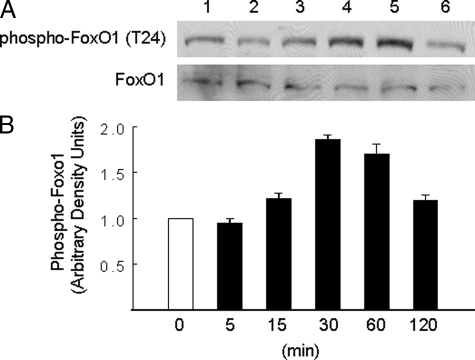
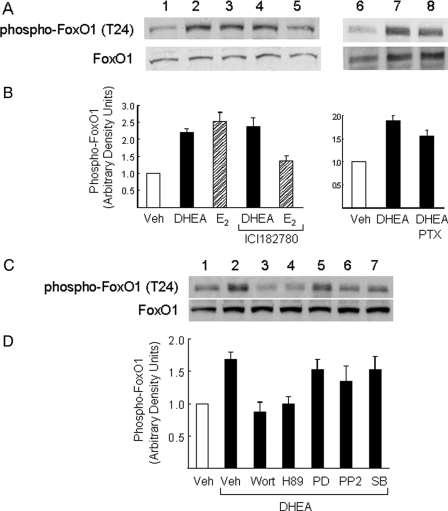
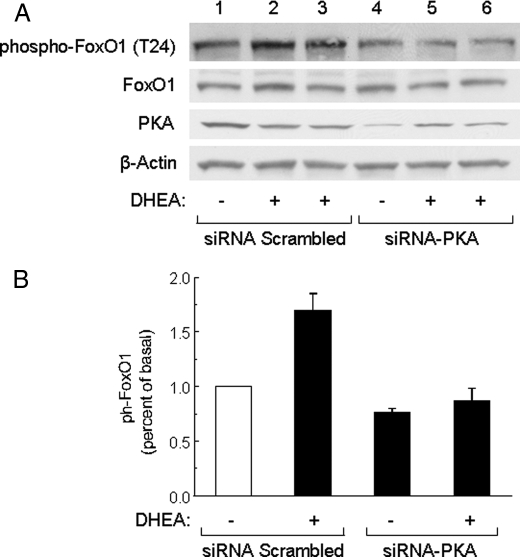
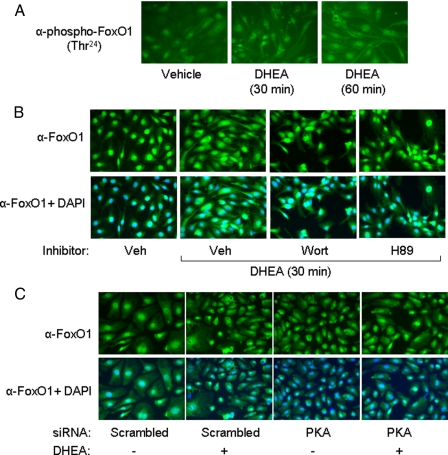
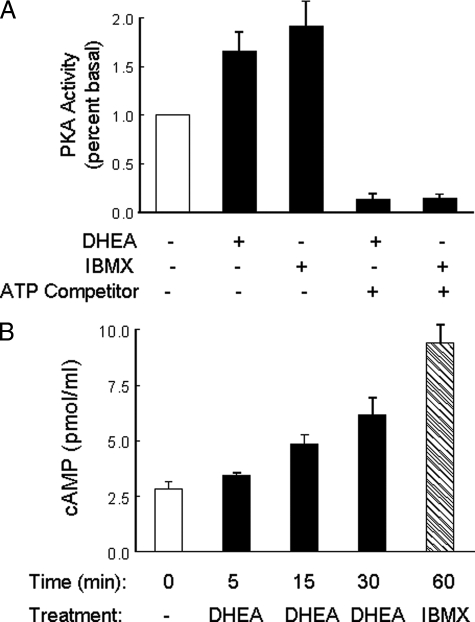
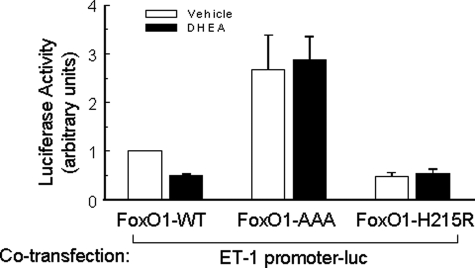
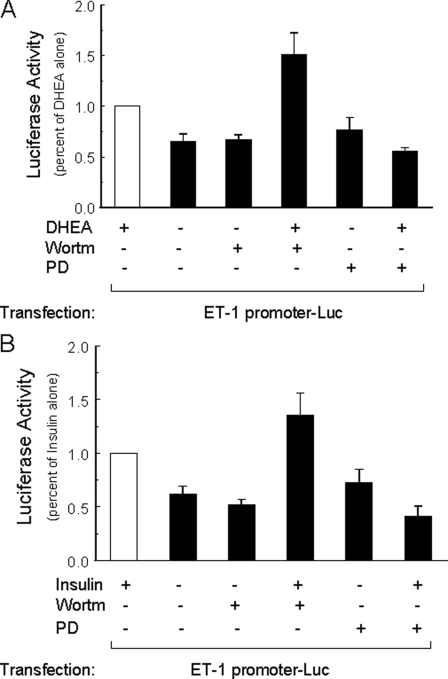
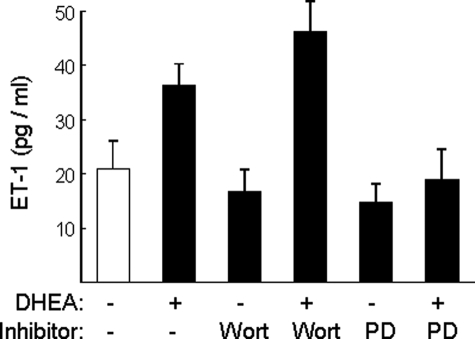
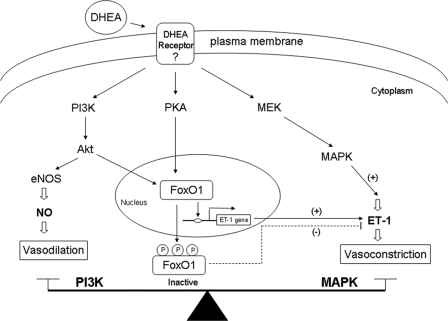
Dehydroepiandrosterone (DHEA) is an endogenous adrenal steroid hormone with controversial actions in humans. We previously reported that DHEA has opposing actions in endothelial cells to stimulate phosphatidylinositol (PI) 3-kinase/Akt/endothelial nitric-oxide synthase leading to increased production of nitric oxide while simultaneously stimulating MAPK-dependent secretion of the vasoconstrictor ET-1. In the present study we hypothesized that DHEA may stimulate PI 3-kinase-dependent phosphorylation of FoxO1 in endothelial cells to help regulate endothelial function. In bovine or human aortic endothelial cells (BAEC and HAEC), treatment with DHEA (100 nM) acutely enhanced phosphorylation of FoxO1. DHEA-stimulated phosphorylation of FoxO1 was inhibited by pretreatment of cells with wortmannin (PI 3-kinase inhibitor) or H89 (protein kinase A (PKA) inhibitor) but not ICI182780 (estrogen receptor blocker), or PD98059 (MEK (MAPK/extracellular signal-regulated kinase kinase) inhibitor). Small interfering RNA knockdown of PKA inhibited DHEA-stimulated phosphorylation of FoxO1. DHEA promoted nuclear exclusion of FoxO1 that was blocked by pretreatment of cells with wortmannin, H89, or by small interfering RNA knockdown of PKA. DHEA treatment of endothelial cells increased PKA activity and intracellular cAMP concentrations. Transfection of BAEC with a constitutively nuclear FoxO1 mutant transactivated a co-transfected ET-1 promoter luciferase reporter. Treatment of BAEC with DHEA inhibited transactivation of the ET-1 promoter reporter in cells overexpressing FoxO1. ET-1 promoter activity and secretion in response to DHEA treatment was augmented by PI 3-kinase blockade and inhibited by MAPK blockade. We conclude that DHEA stimulates phosphorylation of FoxO1 via PI 3-kinase- and PKA-dependent pathways in endothelial cells that negatively regulates ET-1 promoter activity and secretion. Balance between PI 3-kinase-dependent inhibition and MAPK-dependent stimulation of ET-1 secretion in response to DHEA may determine whether DHEA supplementation improves or worsens cardiovascular and metabolic function.
Figures
Similar articles
-
Dehydroepiandrosterone mimics acute actions of insulin to stimulate production of both nitric oxide and endothelin 1 via distinct phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-dependent pathways in vascular endothelium.Mol Endocrinol. 2006 May;20(5):1153-63. doi: 10.1210/me.2005-0266. Epub 2005 Dec 22. Mol Endocrinol. 2006. PMID: 16373398
-
Protein kinase A-alpha directly phosphorylates FoxO1 in vascular endothelial cells to regulate expression of vascular cellular adhesion molecule-1 mRNA.J Biol Chem. 2011 Feb 25;286(8):6423-32. doi: 10.1074/jbc.M110.180661. Epub 2010 Dec 22. J Biol Chem. 2011. PMID: 21177856 Free PMC article.
-
Green tea polyphenol epigallocatechin gallate reduces endothelin-1 expression and secretion in vascular endothelial cells: roles for AMP-activated protein kinase, Akt, and FOXO1.Endocrinology. 2010 Jan;151(1):103-14. doi: 10.1210/en.2009-0997. Epub 2009 Nov 3. Endocrinology. 2010. PMID: 19887561 Free PMC article.
-
Ghrelin has novel vascular actions that mimic PI 3-kinase-dependent actions of insulin to stimulate production of NO from endothelial cells.Am J Physiol Endocrinol Metab. 2007 Mar;292(3):E756-64. doi: 10.1152/ajpendo.00570.2006. Epub 2006 Nov 14. Am J Physiol Endocrinol Metab. 2007. PMID: 17106060
-
Phosphate and Endothelial Function: How Sensing of Elevated Inorganic Phosphate Concentration Generates Signals in Endothelial Cells.Adv Exp Med Biol. 2022;1362:85-98. doi: 10.1007/978-3-030-91623-7_9. Adv Exp Med Biol. 2022. PMID: 35288875 Review.
Cited by
-
Unveiling the metabolic landscape of pulmonary hypertension: insights from metabolomics.Respir Res. 2024 May 28;25(1):221. doi: 10.1186/s12931-024-02775-5. Respir Res. 2024. PMID: 38807129 Free PMC article. Review.
-
Mechanisms for food polyphenols to ameliorate insulin resistance and endothelial dysfunction: therapeutic implications for diabetes and its cardiovascular complications.Am J Physiol Endocrinol Metab. 2013 Sep 15;305(6):E679-86. doi: 10.1152/ajpendo.00377.2013. Epub 2013 Jul 30. Am J Physiol Endocrinol Metab. 2013. PMID: 23900418 Free PMC article. Review.
-
FOXO1 mediates the autocrine effect of endothelin-1 on endothelial cell survival.Mol Endocrinol. 2012 Jul;26(7):1213-24. doi: 10.1210/me.2011-1276. Epub 2012 May 8. Mol Endocrinol. 2012. PMID: 22570335 Free PMC article.
-
Endothelial dysfunction due to selective insulin resistance in vascular endothelium: insights from mechanistic modeling.Am J Physiol Endocrinol Metab. 2020 Sep 1;319(3):E629-E646. doi: 10.1152/ajpendo.00247.2020. Epub 2020 Aug 10. Am J Physiol Endocrinol Metab. 2020. PMID: 32776829 Free PMC article.
-
Bevacizumab Increases Endothelin-1 Production via Forkhead Box Protein O1 in Human Glomerular Microvascular Endothelial Cells In Vitro.Int J Nephrol. 2021 Dec 6;2021:8381115. doi: 10.1155/2021/8381115. eCollection 2021. Int J Nephrol. 2021. PMID: 34912580 Free PMC article.
References
-
- Lasley, B. L., Santoro, N., Randolf, J. F., Gold, E. B., Crawford, S., Weiss, G., McConnell, D. S., and Sowers, M. F. (2002) J. Clin. Endocrinol. Metab. 87 3760-3767 - PubMed
-
- Belanger, A., Candas, B., Dupont, A., Cusan, L., Diamond, P., Gomez, J. L., and Labrie, F. (1994) J. Clin. Endocrinol. Metab. 79 1086-1090 - PubMed
-
- Slowinska-Srzednicka, J., Zgliczynski, S., Soszynski, P., Makowska, A., Zgliczynski, W., Srzednicki, M., Bednarska, M., Chotkowska, E., Woroszylska, M., Ruzyllo, W., and Sadowski, Z. (1991) J. Intern. Med. 230 551-553 - PubMed
-
- Barrett-Connor, E., and Goodman-Gruen, D. (1995) Ann. N. Y. Acad. Sci. 774 259-270 - PubMed
-
- Johannes, C. B., Stellato, R. K., Feldman, H. A., Longcope, C., and McKinlay, J. B. (1999) J. Clin. Epidemiol. 52 95-103 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
