

Regulation of postsynaptic RapGAP SPAR by Polo-like kinase 2 and the SCFbeta-TRCP ubiquitin ligase in hippocampal neurons
- PMID: 18723513
- PMCID: PMC2570879
- DOI: 10.1074/jbc.M802475200
Regulation of postsynaptic RapGAP SPAR by Polo-like kinase 2 and the SCFbeta-TRCP ubiquitin ligase in hippocampal neurons
Abstract
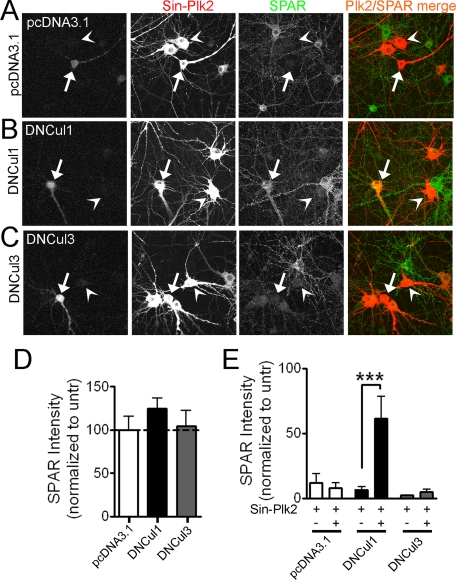
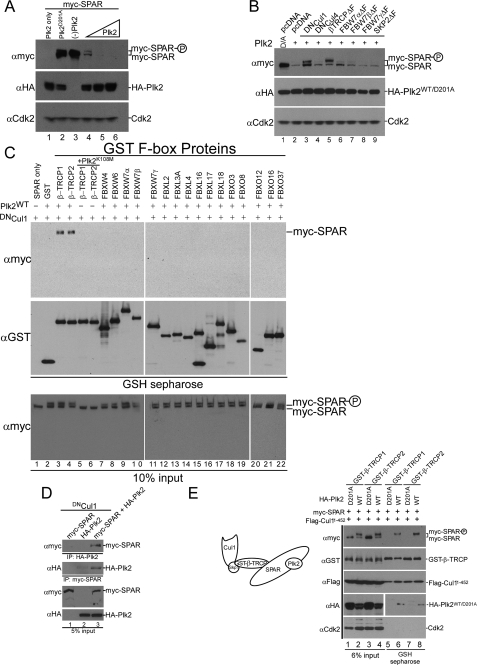
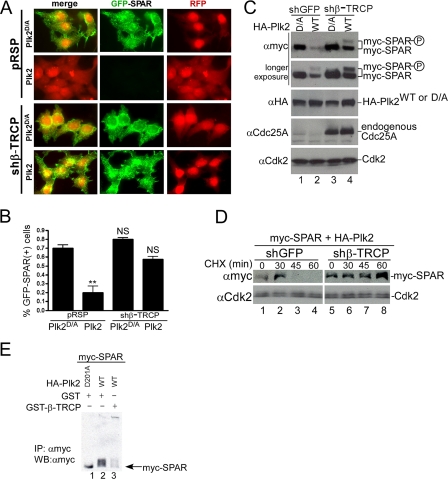
The ubiquitin-proteasome pathway (UPP) regulates synaptic function, but little is known about specific UPP targets and mechanisms in mammalian synapses. We report here that the SCF(beta-TRCP) complex, a multisubunit E3 ubiquitin ligase, targets the postsynaptic spine-associated Rap GTPase activating protein (SPAR) for degradation in neurons. SPAR degradation by SCF(beta-TRCP) depended on the activity-inducible protein kinase Polo-like kinase 2 (Plk2). In the presence of Plk2, SPAR physically associated with the SCF(beta-TRCP) complex through a canonical phosphodegron. In hippocampal neurons, disruption of the SCF(beta-TRCP) complex by overexpression of dominant interfering beta-TRCP or Cul1 constructs prevented Plk2-dependent degradation of SPAR. Our results identify a specific E3 ubiquitin ligase that mediates degradation of a key postsynaptic regulator of synaptic morphology and function.
Figures


References
-
- Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu, N., and Nakahara, H. (2003) Trends Neurosci. 26 360-368 - PubMed
-
- Tada, T., and Sheng, M. (2006) Curr. Opin. Neurobiol. 16 95-101 - PubMed
-
- Bonhoeffer, T., and Yuste, R. (2002) Neuron 35 1019-1027 - PubMed
-
- Carlisle, H. J., and Kennedy, M. B. (2005) Trends Neurosci. 28 182-187 - PubMed
-
- Pak, D. T., Yang, S., Rudolph-Correia, S., Kim, E., and Sheng, M. (2001) Neuron 31 289-303 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
