

RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage
- PMID: 18723675
- PMCID: PMC2529075
- DOI: 10.1073/pnas.0803001105
RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage
Abstract
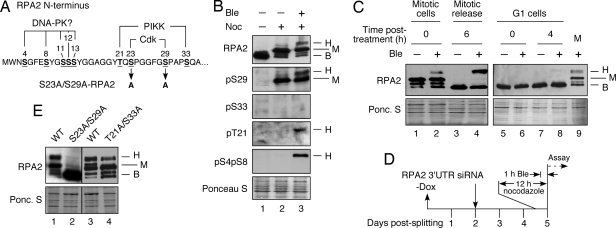
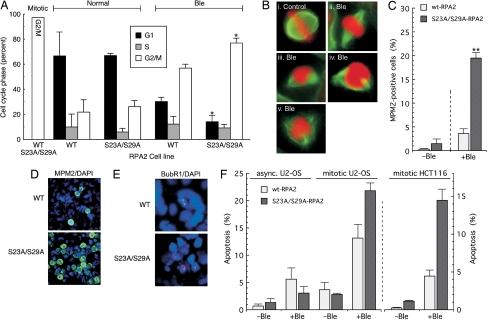
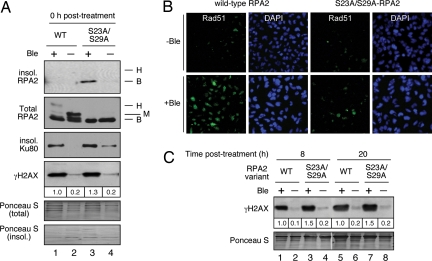
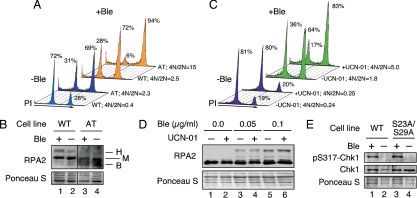
Human replication protein A (RPA) becomes phosphorylated on the RPA2 subunit by cyclin B-Cdc2 during mitosis, although the functional role of this modification is unclear. We find that this modification stimulates RPA2 to become hyperphosphorylated in response to mitotic DNA damage caused by bleomycin treatment. Cells in which endogenous RPA2 was replaced by a mutant subunit lacking both Cdc2 sites had a significant defect in mitotic release into a 2N G(1) phase after exposure to bleomycin. An increased percentage of these mutant cells also was positive initially for cyclin B expression and BubR1 chromatin staining, indicative of an extended spindle assembly checkpoint. The mutant cells that experienced mitotic DNA damage also underwent apoptosis at higher levels than cells expressing the WT subunit. Even so, we did not find the mutation had any dramatic effects on the level of DNA repair in mitosis. Cells lacking ATM (a checkpoint factor and RPA2 kinase) also were severely defective in mitotic exit and were unable to support RPA hyperphosphorylation after mitotic DNA damage. Although checkpoint 1 effector kinase (Chk1) had a more complex role, inhibition of Chk1 activity with UCN-01 also reduced mitotic exit. Chk1 activation and mitotic RPA hyperphosphorylation were found to be independent events. Our results demonstrate that mitotic RPA hyperphosphorylation facilitates release of cells from a damaged mitosis into a 2N G(1) phase, thereby increasing cell viability.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Sanchez Y, et al. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. - PubMed
-
- Skoufias DA, Lacroix FB, Andreassen PR, Wilson L, Margolis RL. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol Cell. 2004;15:977–990. - PubMed
-
- Iftode C, Daniely Y, Borowiec JA. Replication protein A (RPA): The eukaryotic SSB. Crit Rev Biochem Mol Biol. 1999;34:141–180. - PubMed
-
- Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 2004;3:1015–1024. - PubMed
-
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
