

Ether-linked diglycerides inhibit vascular smooth muscle cell growth via decreased MAPK and PI3K/Akt signaling
- PMID: 18723771
- PMCID: PMC2593499
- DOI: 10.1152/ajpheart.00141.2008
Ether-linked diglycerides inhibit vascular smooth muscle cell growth via decreased MAPK and PI3K/Akt signaling
Abstract
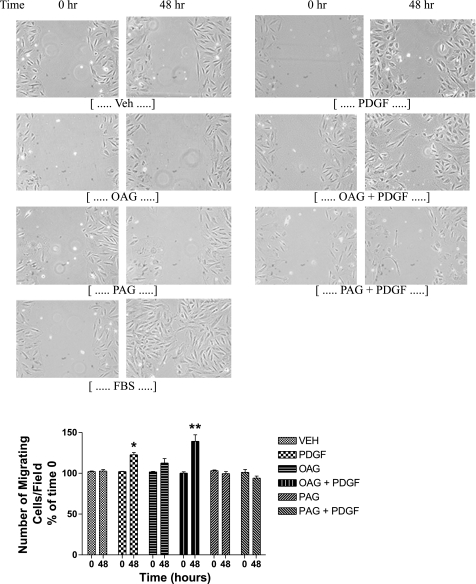
Diglycerides (DGs) are phospholipid-derived second messengers that regulate PKC-dependent signaling pathways. Distinct species of DGs are generated from inflammatory cytokines and growth factors. Growth factors increase diacyl- but not ether-linked DG species, whereas inflammatory cytokines predominately generate alkyl, acyl- and alkenyl, acyl-linked DG species in rat mesenchymal cells. These DG species have been shown to differentially regulate protein kinase C (PKC) isotypes. Ester-linked diacylglycerols activate PKC-epsilon and cellular proliferation in contrast to ether-linked DGs, which lead to growth arrest through the inactivation of PKC-epsilon. It is now hypothesized that ether-linked DGs inhibit mitogenesis through the inactivation of ERK and/or Akt signaling cascades. We demonstrate that cell-permeable ether-linked DGs reduce vascular smooth muscle cell growth by inhibiting platelet-derived growth factor-stimulated ERK in a PKC-epsilon-dependent manner. This inhibition is specific to the ERK pathway, since ether-linked DGs do not affect growth factor-induced activation of other family members of the MAPKs, including p38 MAPK and c-Jun NH(2)-terminal kinases. We also demonstrate that ether-linked DGs reduce prosurvival phosphatidylinositol 3-kinase (PI3K)/Akt signaling, independent of PKC-epsilon, by diminishing an interaction between the subunits of PI3K and not by affecting protein phosphatase 2A or lipid (phosphatase and tensin homologue deleted in chromosome 10) phosphatases. Taken together, our studies identify ether-linked DGs as potential adjuvant therapies to limit vascular smooth muscle migration and mitogenesis in atherosclerotic and restenotic models.
Figures








References
-
- Allen TR, Krueger KD, Hunter WJ 3rd, Agrawal DK. Evidence that insulin-like growth factor-1 requires protein kinase C-epsilon, PI3-kinase and mitogen-activated protein kinase pathways to protect human vascular smooth muscle cells from apoptosis. Immunol Cell Biol 83: 651–667, 2005. - PubMed
-
- Araki S, Tsuna I, Kaji K, Hayashi H. Programmed cell death in response to alkyllysophospholipids in endothelial cells. J Biochem (Tokyo) 115: 245–247, 1994. - PubMed
-
- Barber DF, Alvarado-Kristensson M, Gonzalez-Garcia A, Pulido R, Carrera AC. PTEN regulation, a novel function for the p85 subunit of phosphoinositide 3-kinase. Sci STKE 2006: pe49, 2006. - PubMed
-
- Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J Biol Chem 277: 3286–3292, 2002. - PubMed
-
- Bourbon NA, Yun J, Berkey D, Wang Y, Kester M. Inhibitory actions of ceramide upon PKC-ɛ/ERK interactions. Am J Physiol Cell Physiol 280: C1403–C1411, 2001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous