

Estimation of nucleotide diversity, disequilibrium coefficients, and mutation rates from high-coverage genome-sequencing projects
- PMID: 18725384
- PMCID: PMC2767098
- DOI: 10.1093/molbev/msn185
Estimation of nucleotide diversity, disequilibrium coefficients, and mutation rates from high-coverage genome-sequencing projects
Abstract
Recent advances in sequencing strategies have made it feasible to rapidly obtain high-coverage genomic profiles of single individuals, and soon it will be economically feasible to do so with hundreds to thousands of individuals per population. While offering unprecedented power for the acquisition of population-genetic parameters, these new methods also introduce a number of challenges, most notably the need to account for the binomial sampling of parental alleles at individual nucleotide sites and to eliminate bias from various sources of sequence errors. To minimize the effects of both problems, methods are developed for generating nearly unbiased and minimum-sampling-variance estimates of a number of key parameters, including the average nucleotide heterozygosity and its variance among sites, the pattern of decomposition of linkage disequilibrium with physical distance, and the rate and molecular spectrum of spontaneously arising mutations. These methods provide a general platform for the efficient utilization of data from population-genomic surveys, while also providing guidance for the optimal design of such studies.
Figures
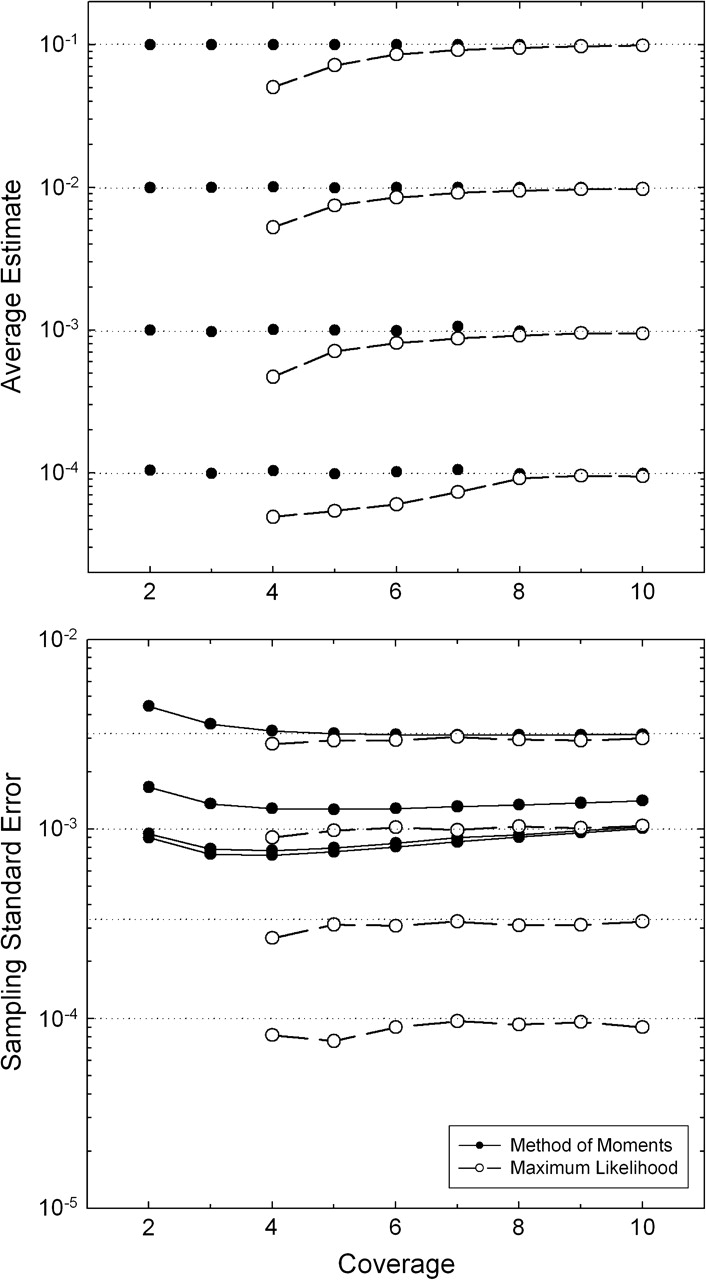
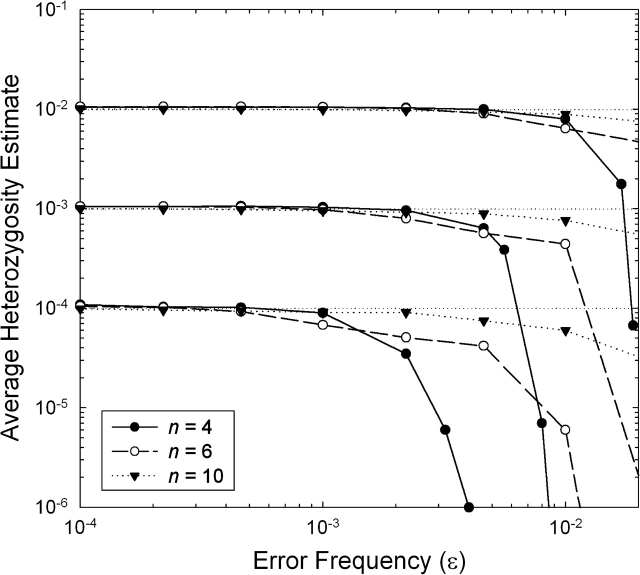
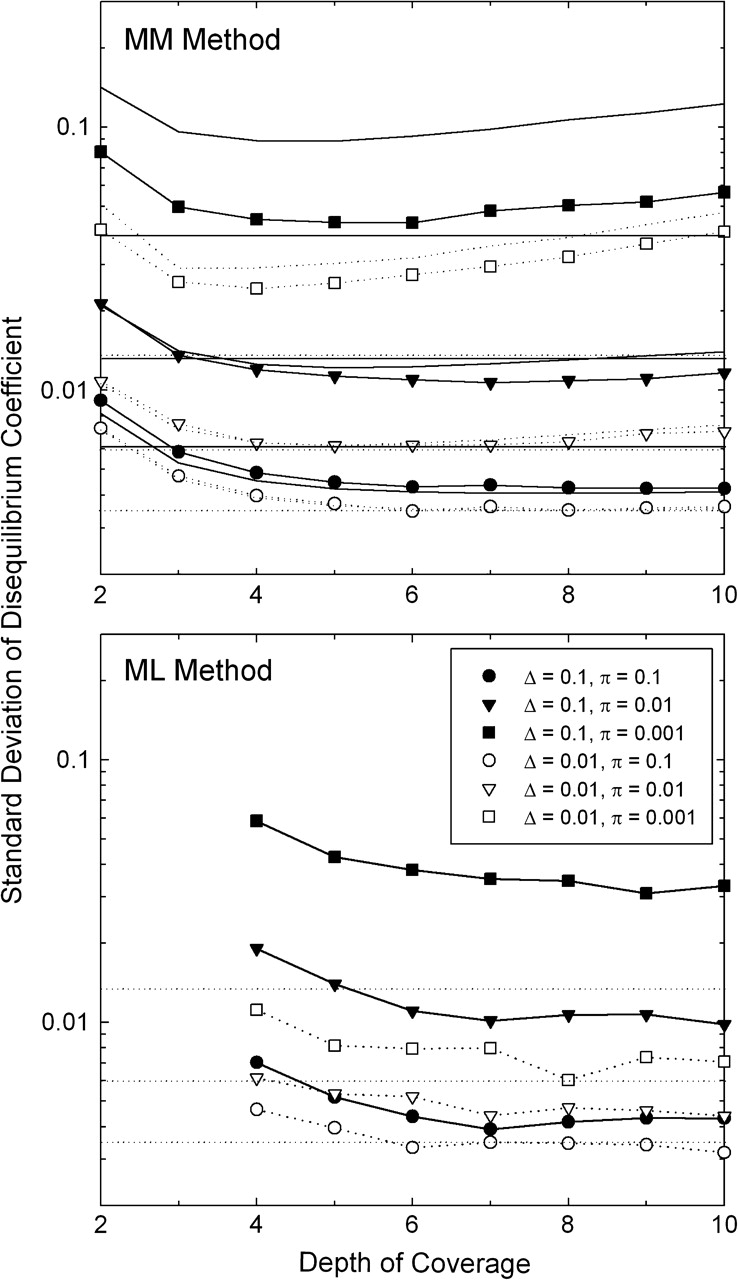
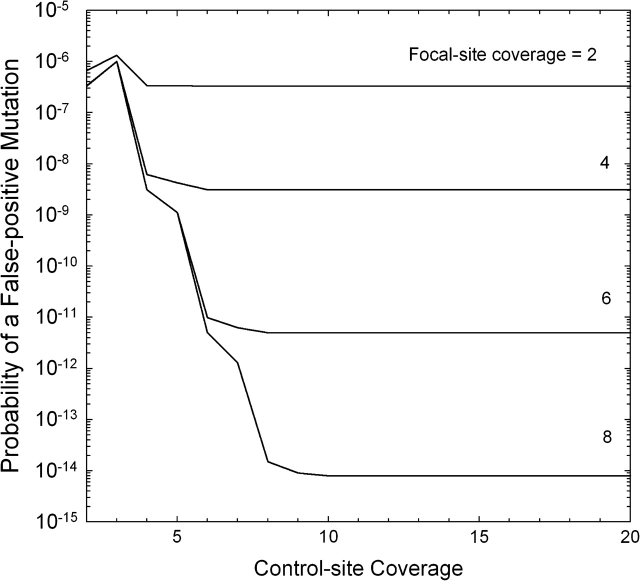
References
-
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. - PubMed
-
- Clark AG, Whittam TS. Sequencing errors and molecular evolutionary analysis. Mol Biol Evol. 1992;9:744–752. - PubMed
-
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. - PubMed
-
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
