

Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation
- PMID: 18725990
- PMCID: PMC2518075
- DOI: 10.1172/JCI35424
Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation
Abstract
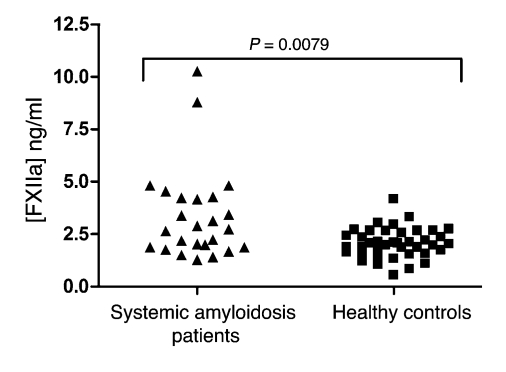
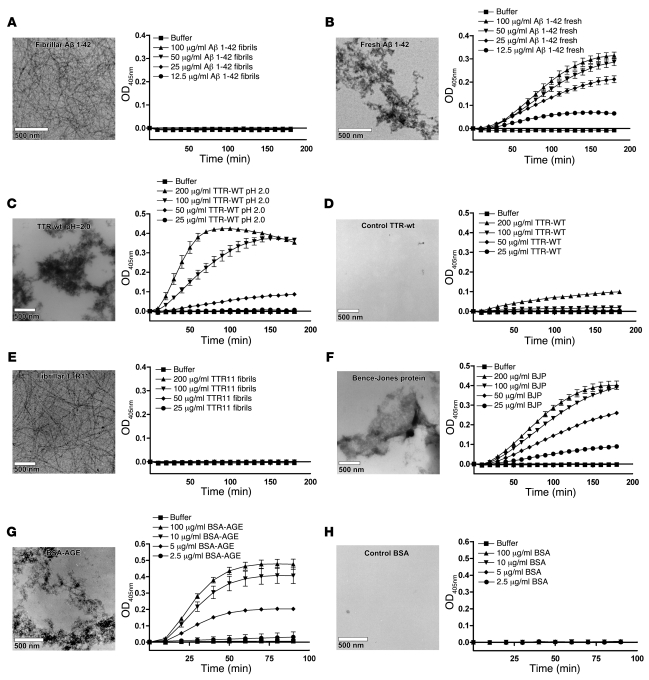
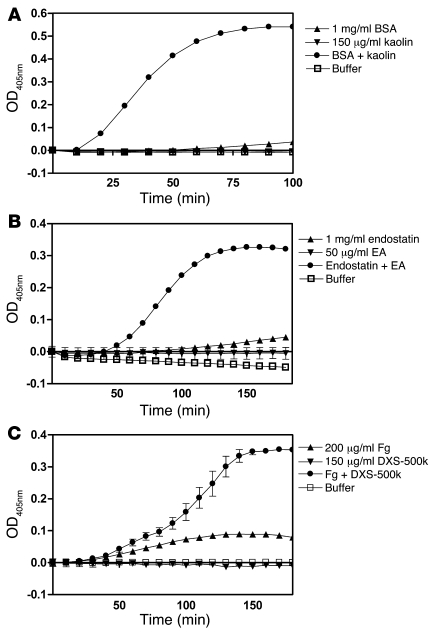
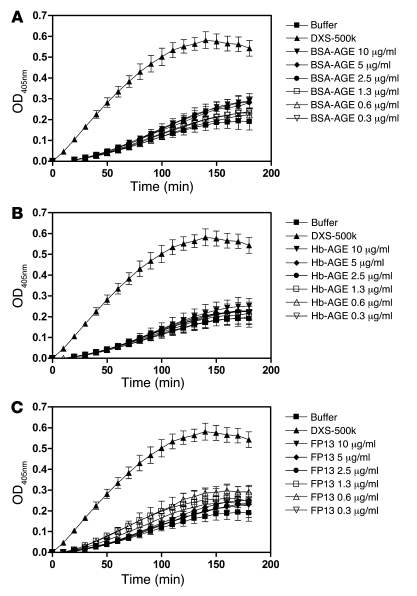
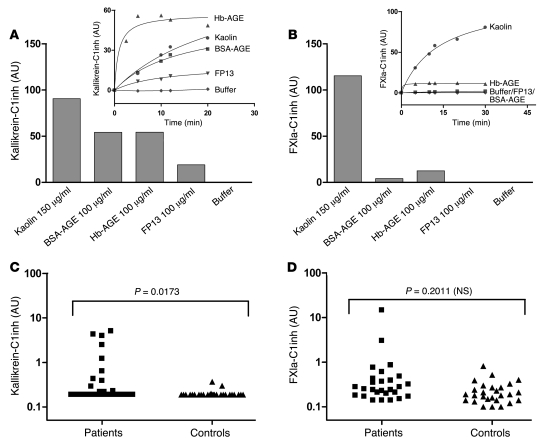
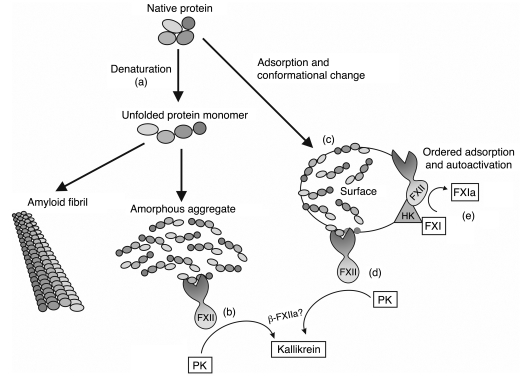
When blood is exposed to negatively charged surface materials such as glass, an enzymatic cascade known as the contact system becomes activated. This cascade is initiated by autoactivation of Factor XII and leads to both coagulation (via Factor XI) and an inflammatory response (via the kallikrein-kinin system). However, while Factor XII is important for coagulation in vitro, it is not important for physiological hemostasis, so the physiological role of the contact system remains elusive. Using patient blood samples and isolated proteins, we identified a novel class of Factor XII activators. Factor XII was activated by misfolded protein aggregates that formed by denaturation or by surface adsorption, which specifically led to the activation of the kallikrein-kinin system without inducing coagulation. Consistent with this, we found that Factor XII, but not Factor XI, was activated and kallikrein was formed in blood from patients with systemic amyloidosis, a disease marked by the accumulation and deposition of misfolded plasma proteins. These results show that the kallikrein-kinin system can be activated by Factor XII, in a process separate from the coagulation cascade, and point to a protective role for Factor XII following activation by misfolded protein aggregates.
Figures
Comment in
-
The elusive physiologic role of Factor XII.J Clin Invest. 2008 Sep;118(9):3006-9. doi: 10.1172/JCI36617. J Clin Invest. 2008. PMID: 18725991 Free PMC article. Review.
References
-
- Bouma B.N., von dem Borne P.A., Meijers J.C. Factor XI and protection of the fibrin clot against lysis — a role for the intrinsic pathway of coagulation in fibrinolysis. Thromb. Haemost. 1998;80:24–27. - PubMed
-
- Naito K., Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J. Biol. Chem. 1991;266:7353–7358. - PubMed
-
- Forbes C.D., Pensky J., Ratnoff O.D. Inhibition of activated Hageman factor and activated plasma thromboplastin antecedent by purified serum C1 inactivator. J. Lab. Clin. Med. 1970;76:809–815. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
