

The treatment of hyperhomocysteinemia
- PMID: 18729731
- PMCID: PMC2716415
- DOI: 10.1146/annurev.med.60.041807.123308
The treatment of hyperhomocysteinemia
Abstract
The unique biochemical profile of homocysteine is characterized by chemical reactivity supporting a wide range of molecular effects and by a tendency to promote oxidant stress-induced cellular toxicity. Numerous epidemiological reports have established hyperhomocysteinemia as an independent risk factor for cardiovascular disease, cerebrovascular disease, dementia-type disorders, and osteoporosis-associated fractures. Although combined folic acid and B-vitamin therapy substantially reduces homocysteine levels, results from randomized placebo-controlled clinical trials testing the effect of vitamin therapy on outcome in these diseases have generally fallen short of expectations. These results have led some to abandon homocysteine monitoring in the management of patients with cardiovascular or cognitive disorders. These trials, however, have generally included patients with only mildly elevated homocysteine levels and have not addressed several clinical scenarios in which homocysteine reduction may be effective, including the primary prevention of atherothrombotic disease in individuals at low or intermediate risk, or those with severe hyperhomocysteinemia.
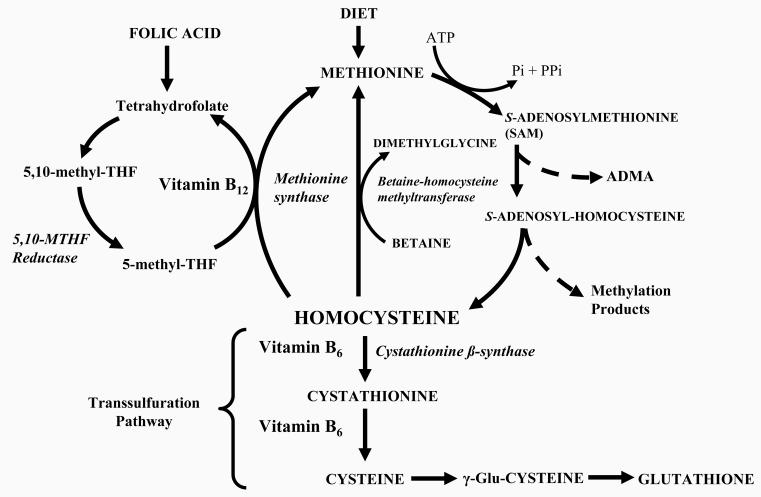
Figures


References
-
- Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. J. Am. Med. Assoc. 1995;74:1049–57. - PubMed
-
- Homocysteine Studies Collaboration Homocysteine and risk of ischemic heart disease and stroke; a meta-analysis. J. Am. Med. Assoc. 2002;288:2015–22. - PubMed
-
- Handy DE, Zhang Y, Loscalzo J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J. Biol. Chem. 2005;280:15518–25. - PubMed
-
- Zhou J, Werstuck GH, Lhoták S, de Koning AB, Sood SK, et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–13. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
