

Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line
- PMID: 18753622
- PMCID: PMC2529074
- DOI: 10.1073/pnas.0806437105
Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line
Abstract
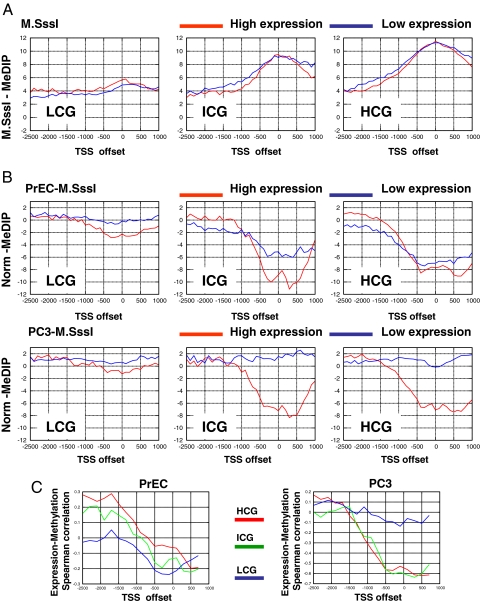
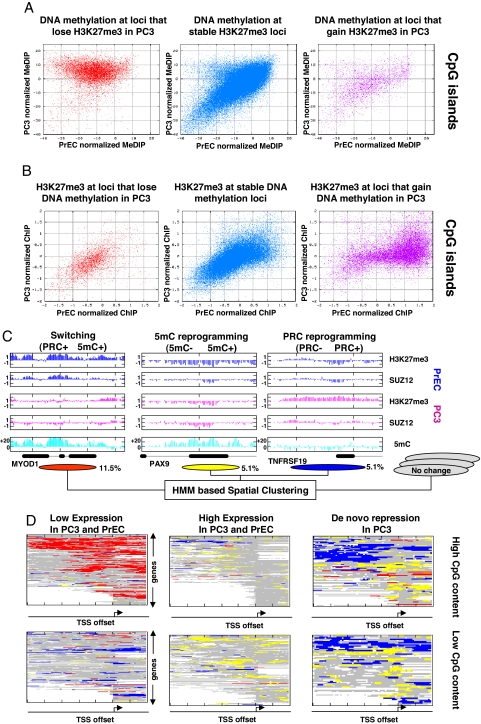
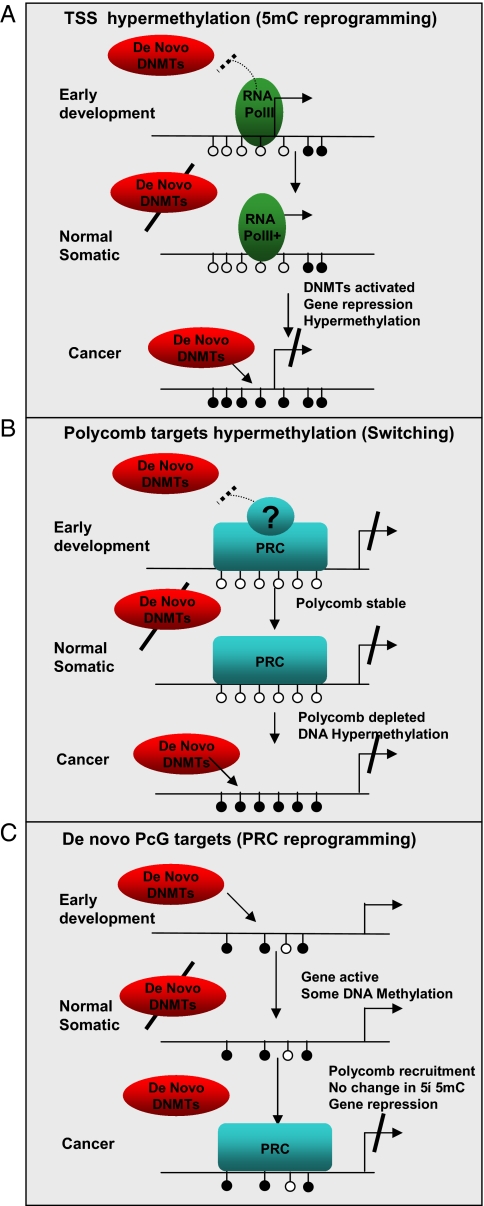
Epigenetic reprogramming is commonly observed in cancer, and is hypothesized to involve multiple mechanisms, including DNA methylation and Polycomb repressive complexes (PRCs). Here we devise a new experimental and analytical strategy using customized high-density tiling arrays to investigate coordinated patterns of gene expression, DNA methylation, and Polycomb marks which differentiate prostate cancer cells from their normal counterparts. Three major changes in the epigenomic landscape distinguish the two cell types. Developmentally significant genes containing CpG islands which are silenced by PRCs in the normal cells acquire DNA methylation silencing and lose their PRC marks (epigenetic switching). Because these genes are normally silent this switch does not cause de novo repression but might significantly reduce epigenetic plasticity. Two other groups of genes are silenced by either de novo DNA methylation without PRC occupancy (5mC reprogramming) or by de novo PRC occupancy without DNA methylation (PRC reprogramming). Our data suggest that the two silencing mechanisms act in parallel to reprogram the cancer epigenome and that DNA hypermethylation may replace Polycomb-based repression near key regulatory genes, possibly reducing their regulatory plasticity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
-
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. - PubMed
-
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
